
Abstract
Tackling the challenges of chemical exposomics will require the implementation of diverse analytical strategies and technological advancements. Herein, high-resolution mass spectrometry-based methods applied in current chemical exposome studies have been surveyed and are shown to be limited. Notably, liquid chromatography separations almost exclusively employ reversed-phase C18 columns using water/methanol gradients with formic acid additive, while gas chromatography is underexploited in the field at this stage. A systematic evaluation of strategies applied in related disciplines (i.e. metabolomics, proteomics, multiresidue trace analysis) was undertaken to provide practical guidance for the development of chemical exposomics. The approaches were assessed on the basis of their costs (i.e. capital expenditure, overhead and maintenance fees, expertise required, consumables) and potential benefits (i.e. improvements to sensitivity, coverage, reproducibility, throughput, ease of use) to prioritize those with promise for chemical exposomics application. Alongside a need for technological investments (e.g. advanced hardware updates), numerous low-cost strategies showed high potential benefits (e.g. different column phases, enhanced sample fractionation) and are feasible for rapid adoption.
INTRODUCTION
Coupling the comprehensive analysis of chemical exposures (i.e. chemical exposomics) with other omics (i.e. metabolomics, proteomics) is a promising strategy for linking chemical exposures to specific biological responses,1-3 chemical prioritization2,4 and to lead exposure-based risk assessment.3,5–7 The profiling of small endogenous compounds and exogenous chemicals converge in terms of platforms used (e.g. high-resolution mass spectrometry [HRMS]8–12), m/z range covered3,13 and the analytical challenges faced, such as the need to increase coverage (i.e. physicochemical property range) and sensitivity. These challenges are amplified when profiling trace analytes and if working with low sample volumes/amount of complex matrix.14–16 Despite approaches to enhance the analytical sensitivity and coverage of endogenous compounds also having the potential to improve the detection of exogenous compounds,17 many remain underexploited for the profiling of chemical exposure agents, and further exchange between the fields of chemical exposomics and metabolomics should be encouraged.11 Additionally, multiresidue methods to assay hundreds to thousands of target contaminants in complex matrices (e.g. food/plant) have recently been developed.18–21 Although these approaches are selective for known chemicals, the breadth of chemicals assayed means sample preparation and chromatography is akin to profiling, and evidence that large-scale quantification of low abundant chemicals is feasible.
We have reviewed the gas chromatography (GC)-HRMS and liquid chromatography (LC)-HRMS approaches applied in chemical exposome studies and evaluated strategies used in related disciplines which may enhance analyte detection, coverage, sample throughput, and reproducibility. The strategies that warrant further investigation have been prioritized through an assessment of their relative costs and potential benefits of implementation in chemical exposomics. The review and assessment were restricted to GC- and LC-HRMS workflows, because their use is most widespread, but we guide readers to reviews of alternative platforms that provide appealing features for chemical exposomics, e.g. ion mobility-mass spectrometry22 and supercritical fluid chromatography.23 In this article, we focus on the laboratory-based components and do not venture into the range of bioinformatic and computational approaches necessary for data analysis.
The work is articulated in 3 parts:
Analytical methodologies used for HRMS-based chemical exposomics were compiled from literature with the purpose to identify the most commonly applied approaches and pinpoint lacks and areas for improvement. From this survey, a clear trend emerged: rather uniform analytical conditions are applied for HRMS-based investigation of the chemical exposome.
Since the approaches applied in chemical exposomics are limited, we reviewed strategies used in related disciplines to identify those with potential for advancing chemical exposome research. Particular attention was paid to those applied in metabolomics because of the analytical and conceptual similarities with chemical exposomics,24,25 and applications from proteomics were considered. Targeted analysis of environmental contaminants was also considered if a broad range of compounds in complex matrix were assayed.
The costs and benefits of the reviewed strategies were assessed and evaluated with the aim to prioritize approaches with potential for application in chemical exposomics studies. Focus was upon strategies that could be adopted in the near future. The evaluation was based on literature findings and authors’ experiences and a summarized perspective is presented.
Analytical approaches currently used for chemical exposomics are rather uniform
Literature was surveyed during December 2020 to identify analytical approaches applied in HRMS-based chemical exposomics. The keywords/phrases “non target analysis” (title), “non target analysis environmental chemicals” (topic), “suspect screening” (title), and “exposome” (topic) were used to search Web of Science.
Whereas targeted analysis methods selectively measure chemicals of already confirmed identity, non-target analysis, and suspect screening are typically performed with the MS scanning a wide m/z range to enable broad-scope chemical profiling.8,26,27 Non-target analysis aims to detect features without any a priori information while suspect screening relies on some a priori considerations, seeking to detect chemicals expected to be present in a sample.8,26,27 Unlike target analysis, non-target analysis and suspect screening provide a putative identification that requires subsequent efforts for confirmation.
The survey was a formal search of studies presented as chemical exposomics and suspect screening/non-target analyses of environmental chemicals in biological and/or environmental matrices.
It is acknowledged that the selected terms are not comprehensive but the survey is intended to provide an overview of the trends of recent studies in the field. The retrieved manuscripts were manually inspected by scientists with the appropriate expertise to identify studies that employed suspect screening/non-target workflows. In total, 144 publications reporting full-scan HRMS-based analysis were selected and compiled (see Supplementary Data). From this survey, it emerged that experimental conditions commonly used in chemical exposomics are rather uniform.
Solvent extraction and solid-phase extraction (SPE) are the most frequently used techniques for sample extraction of human matrices28 and the same trend is evident when extending to other (non-human) biological specimens (Tables S1.1 and S2.1). This was to be expected because minimally selective sample preparation/extraction methods are often advocated for chemical exposomics.27–29 Notably, no study applied pre-normalization procedures to correct for matrix effects or to reduce the dynamic range of concentration (e.g. specific gravity, creatinine normalization).
Across all publications, LC is the most commonly used separation platform compared to GC: 172 LC-HRMS methods were reported for LC-HRMS (28 applied to biospecimens, 126 to other matrices, 18 to both; Table S3.1) while 29 methods were reported for GC-HRMS (9 applied to biospecimens, 18 to other matrices, 1 applied to both; Table S4.1).
The majority of LC studies used electrospray ionization (ESI), with acquisitions performed in both positive and negative polarity while atmospheric pressure chemical ionization (APCI) had limited application (Table S3.1). Reversed-phase (RP) chromatography was commonly performed by using C18-based columns and water/methanol gradients, with formic acid, at temperatures below 50 °C (Tables 1 and S3.1). Column lengths and particle diameters varied from 50 to 150 mm and 1.7-3.5 µm, respectively, while an internal diameter of 2.1 mm was common (Tables 1 and S3.1).
LC Conditions commonly used in suspect screening and non-target studies
| Ionization | Particle chemistry | Column ID | Mobile phase | Modifier | Oven temperature |
|---|---|---|---|---|---|
| ESI+ | C18-based | narrow bore | H2O/MeOH or ACN | FA or FA + AF/AA | <50°C |
| (84%)a (n = 137) | (81%) (n = 135) | (95%)b (n = 136) | (75%)c (n = 132) | (97%) (n = 89) | |
| ESI − | C18-based | narrow bore | H2O/MeOH or ACN | FA or FA + AF/AA | <50°C |
| (86%)d (n = 116) | (76%) (n = 114) | (94%)e (n = 115) | (48%)f (n = 108) | (90%) (n = 72) |
FA: Formic acid. AF: Ammonium formate. AA: Ammonium acetate. MeOH: methanol. ACN: acetonitrile. a59% C18; 25% C18 modified for polar compound retention/hybrid phases. b55% H2O/MeOH; 40% H2O/ACN. c59% FA; 16% FA + AF/AA. d59% C18; 27% C18 modified for polar compound retention/hybrid phases. e57% H2O/MeOH; 37% H2O/ACN. f41% FA; 7% FA + AF/AA.
Among the limited applications of GC separation, 5-type columns were generally used (Table S4.1) coupled with electron ionization (EI), operated in positive acquisition mode at 70 eV (Table S4.1).
The lack of versatility/diversity of current HRMS analytical approaches was recently highlighted as an obstacle limiting the comprehensive characterization of the internal chemical exposome.24 It was evidenced that HRMS-based methodologies have limited capability to detect the wide range of environmental pollutants and consumer chemicals in human blood18; due to their abundance typically being orders of magnitude lower than many endogenous compounds and drugs/pharmaceuticals.24,30–32 Furthermore, the dynamic range of chemical concentrations in samples far exceeds that of MS detectors, meaning that multiple different extractions and enrichments will be essential for comprehensive detection. The diversification of methodologies must also be balanced with achieving highly reproducible workflows that provide a solid basis for harmonizing procedures across multiple laboratories to generate comparable data at large scale.
Analytical strategies to diversity chemical exposomics looking across related fields
Sample treatment
Pre-analytical procedures
Sample storage and stability
Sample storage, stability, and handling are crucial variables affecting analytical outcomes. The impact of pre-analytical procedures on coverage of endogenous metabolites has been comprehensively reviewed33–35 and guidelines for biological specimens elaborated.36,37 Chemical lability has been of minor concern for the measurement of exogenous compounds because the traditional chemicals of focus are persistent (e.g. persistent organic chemicals) or relatively stable (e.g. pesticides). These chemicals could potentially be measured many (tens of) years after sample collection with minimal provisions undertaken for storage. Yet, more comprehensive characterization of the chemical exposome extends the focus toward more polar and labile contaminants.38 Cohort studies often involve long storage time (tens of years) and implementing greater protective measures for storage would enable the prospective integration with assays of less stable molecules (e.g. proteins). Hebels et al. reported that so long as consistent procedures have been implemented, biobank samples (stored at –80°C) would be suitable for meaningful multiomics analysis even after long storage periods (∼15 years).39
Studies comparing the long-term impact of different storage conditions are limited but some generic measures beneficial to preserve sample integrity include (1) quenching (e.g. dry ice, liquid nitrogen) immediately after sample collection and appropriate sample sealing to prevent losses of volatile compounds40; (2) storage at −80°C or −196°C using liquid nitrogen, to prevent biotransformation reactions and prevent potential losses41,42; (3) limiting storage time and frosting/defrosting cycles (e.g. by aliquoting subsamples in advance) and minimizing sample handling.40,42
Further investigations to assess the long-term impacts of storage conditions on chemical components of biospecimen and environmental matrices are encouraged, and are especially critical for large-scale studies, where the analyzed samples have often been stored for different periods of time. The diversity of intracellular biochemical reactions that exogenously-derived chemicals are involved is typically less than for endogenous compounds43 which facilitates comparatively more accurate prediction of biotransformation and modeling of chemical exposure than for metabolic networks. Approaches similar to the ones used to estimate chemical biodegradation could be attempted to model biotransformation reactions in biospecimens over storage. For example, Quantitative Structure-Biodegradability Relationships (QSBRs) are widely used to predict chemical biodegradability by using molecular descriptors.44 Time and cost-efficient approaches to extrapolate half-lives reading across different environmental media have been recently proposed for small molecules, where activated sludges were used as accelerated reactors to calculate half-lives of various agrochemicals and regression models developed to extrapolate to soil half-lives.45 Similarly, in vitro “reactors” with biological extracts enriched with degradative molecules and enzymes or genetically modified microorganisms (e.g. to over-express cytochrome P450 enzymes) could be used to provide biotransformation rates of chemicals in biological specimens to eventually extrapolate with temperature corrections (e.g. by Arrhenius equation)46 or correlations with measurements at lower temperatures.
Sample normalization
To reduce variability across biological samples and obtain comparable data, sample normalization is fundamental, especially when approaching quantification.47,48 Otherwise, the correct interpretation of the results can be confounded by large inter-individual and intra-individual variation. For example, dilution factors for urine may vary up to a factor of 15,49 leading to different detection limits and potential matrix interferences. For more details on normalization strategies for biological specimens (e.g. protein/lipid normalization, specific gravity/osmolarity, weight/volume base methods), numerous reviews are available.34,47–49
Pre-analytical normalization is more relevant for exocrine-related glands/tissues/fluids (e.g. urine, kidney, gastrointestinal tract) compared to endocrine (e.g. blood, thyroid, ovaries, etc.); a consequence of the role in maintaining homeodynamic regulation. High variability limits comparative analysis of relative abundances due to varying limits of detection, and is further complicated, e.g. by signal ratio bias from non-linear ESI responses. This is particularly problematic for low abundant features because it reduces detection frequencies and sparsity cannot easily be corrected post-acquisition. On the other side, pre-analytical procedures to minimize signal variability typically involve dilution of samples, reducing the overall number of compounds detectable. Analyzing serial dilutions of representative samples to assess potential matrix effects by evaluating response linearity was recently recommended over procedures with extensive sample manipulation, with direct injection demonstrated to provide more reliable semi-quantification in many cases.50,51
Post-analytical normalization strategies by using dedicated software and statistical approaches can be used to correct for sensitivity drifts and reduce variability across samples.48 However, exogenously-derived compounds are present at lower abundance and with reduced frequency compared to constitutive metabolites and proteins, limiting applicability and validity of typical corrective procedures (e.g. noise thresholds and detection frequency filters, missing value imputation).52
Longitudinal studies become even more complex because analytical batch variation is often larger than temporal changes, so randomizing measurements may not be enough to ensure data comparability. In addition, at the point of analysis, it is common that samples have been stored for varying lengths of time, adding further complexity. Consequently, the implementation of normalization procedures requires careful consideration for each chemical exposomics study and more long-term storage studies would also help to advance identification of suitable normalization parameters.
Sample extraction and purification
Finding the best compromise between selectivity and sensitivity can be challenging and careful consideration is needed with respect to the specific purposes of the study.27 Sample homogenization is critical to obtain consistent extraction efficiency, especially for heterogenous matrices (e.g. tissues, fecal samples, cell cultures).53 Sample clean-up can reduce recoveries for compounds of interest but greater removal of interferences and reduced matrix effects can enable increased concentration of extracts, providing better sensitivity to compensate losses.54
Profiling sub-exposome components: partitioning/fractionation and selective recognition
Sample fractionation has been highlighted as a potential approach for the in-depth characterization of biospecimen because greater enrichment factors can be achieved through concentration of fractions and a combination of platforms can be used for more focused analysis of different fractions.24 However, each stage of purification entails losses and the initial sample amount available is the main limiting factor.
Solvent partitions allow for the separation of analytes in different fractions based on their hydrophobicity that can be processed separately or purified and then pooled together. For example, Pourchet et al. proposed a non-targeted workflow for investigating halogenated compounds in human milk after protein and lipid removal by using solvent partition to separate analytes in two different fractions for GC-HRMS and LC-HRMS analysis.55 Furthermore, hot solvent mixtures,14 microwave,56 and ultrasound57 can be used to enhance solvent extraction efficiency. SPE-based fractionations, via sequential elution58,59 or the use of sequential phases,60 have been evidenced to increase metabolome coverage61 and can equally be applied for enhancing coverage of the chemical exposome, capitalizing on the wide range of SPE sorbents are commercially available. For example, step-wise elution from mixed-mode SPE was used to separate neutral and acidic polycyclic aromatic compounds in runoff water samples and fractions screened via GC- and LC-MS, uncovering numerous emerging biotransformation products,62 while a two-stage hydrophilic–lipophilic balance (HLB)-activated carbon SPE purification was applied to screen hundreds of chemicals in waters.63 Plates and cartridges provide an alternative to the traditional precipitation techniques for removing proteins and lipids.64,65 For example, a phospholipid depletion followed by cation exchange SPE enabled greater concentration of plasma sample extracts prior to instrumental analysis compared to classical protein precipitation, showing increased sensitivity.54
The use of molecularly imprinted polymers (MIPs) represents a promising approach to selectively purify specific structural classes of compounds, e.g. halogenated,66 organophosphate,67 and glucuronidated68 chemicals, and shows greater cleanup efficiency to enable enhanced concentration of low abundant analytes.69 Similarly, chemoselective probes that consist of a solid support containing a probe with specific sites to bind target groups and cleavage linkers to release the compound in certain conditions70 have been successfully applied for enrichment in metabolite profiling studies,70,71 e.g. targeting amines,72 aldehydes, and ketones.73 Protein receptors can be used as probes for selection of biologically-active chemicals74 and multiple MIPs and/or probes can be used in tandem to enable highly resolved fractionation. However, applications are limited by the lack of commercially available chemistries.
Chemical and enzymatic treatments
Chemical reactions (e.g. deconjugation, derivatization) can promote analyte release from matrix components and/or enhance ionization efficiency. Deconjugation has been commonly used in target assays (e.g. for bisphenols) to measure the total concentration of the parent compounds75 and recommended for suspect screening to confirm the identity of parent substances by comparing non-deconjugated and deconjugated samples.76 Similar comparative profiling has also been conducted using glucuronidase and sulfatase with high activity and promiscuity for non-target analysis of glucuronide77 and sulfate metabolites,78 respectively, greatly increasing analyte detection. Adoption in chemical exposomics studies would advance toward more comprehensive screening of Phase 2 detoxification metabolism. However, obtaining such high activity requires multistep purification from commercial enzyme extracts, thus requiring a great level of expertise. Alternatively, typically more affordable chemical deconjugations could be used but their decreased specificity complicates annotation/identification.
Different reaction rates mean it is difficult to obtain complete deconjugation for a wide range of substances79 and challenges to standardize procedures and reaction conditions often leads to large intra- and inter-lab variability.27 Furthermore, unexpected transformation products and degradation of parent compounds can occur80 and information of biotransformation patterns can be lost, which is detrimental for fate and toxicity assessment across the chemical life cycle.
Besides deconjugation, enzymes can be used to assist extraction by degrading cell/membranes and matrix components.81 For example, Wawrzyniak et al. applied a mild proteinase K digestion to human plasma to misfold proteins and induce the release of associated metabolites.82 The method increased metabolome coverage compared to direct precipitation, more than doubling the number of annotated, reproducible features, though the formation of non-specific protein fragments was also noted. Application of a protease treatment step could also be beneficial for the enhanced detection of environmental chemicals since many are known to associate with proteins, such as per- and poly-fluorinated substances.83 Similarly, the potential of other biological enzymes (e.g. lipases) could be explored as milder digestion alternatives to traditional strong acid/alkali treatments often employed.84
Chemical derivatization can be used to make compounds more volatile for GC,85 extend the hydrophobicity range covered by LC (e.g. for better separation of very polar compounds)86 and increase detection sensitivity through the addition of moieties that improve ionization, e.g. halogens or alkyl groups. Various derivatization agents can be combined to target diverse groups of analytes87 and improve confidence for annotation (e.g. comparing labeled and non labeled sample fractions/features), although libraries will need to be extended to include more derivatized compounds.88 For example, Zhao et al.87 proposed a 4-channel chemical isotope labeling method to quantify hydroxyls, amines/phenols, carboxylic acids, and ketones/aldehydes. However, optimization of reaction for each sub-class is complex. The slow reaction speeds can limit throughput for use of some derivatization agents86 and the potential instability of derivates makes it challenging to ensure reproducibility without automatization.89,90 Although usually performed in the latest stage of sample preparation,86 derivatization before sample extraction can be used to increase the affinity of polar compounds toward polymeric phases (e.g. SPE cartridges)91 or solvents. The potential of this approach has been demonstrated for assessment of the urinary chemical exposome, with enhanced detection sensitivities from 2- to 1184-fold.92
Microextraction techniques and passive sampling
The main microextraction techniques can be classified as solved-based i.e. liquid-phase microextraction (LPME), or sorbent-based, i.e. solid-phase microextraction (SPME).93 LPME consists liquid–liquid extractions using small solvent volumes, with the most common approaches being (1) dispersive liquid–liquid microextraction (DLLME), where droplets of a non-polar solvent are finely dispersed in an aqueous sample allowing for a rapid extraction; (2) hollow fiber-protected liquid-phase microextraction (HF-LPME), where analytes are extracted by a solvent placed in the pores of a hollow fiber and then transferred in an acceptor phase and (3) electromembrane microextraction (EME), which is similar to HF-LPME but supported by the application an electrical field. LPME present numerous advantages, such as reduced solvent consumption, low cost, broad selectivity, and rapidity. However, working with such small volume extracts can pose challenges for automation. More details on these techniques and applications can be found in recent reviews94,95 and their potential to improve selectivity and efficiency in sample clean-up should be further investigated for chemical exposomics. The advantages of DLLME for broad extraction for multiple novel psychoactive compounds have been shown.96
SPME has been more widely applied in metabolomics research,40,97 often showing performance comparable or even better than traditional approaches.98–101 Non-exhaustive extraction methods, such as SPME, can provide better sensitivity for low abundant species since suppression from matrix components is reduced.98,99 Comparatively, a limited number of studies have been published on non-target analysis of environmental chemicals using passive sampling approaches,102–107 but applications in targeted studies show their potential for concentrating low abundant chemicals from complex matrixes (e.g. hydrophobic organic chemicals in waters or soil/sediment porewaters).108,109 Passive sampling (e.g. by SPME, silicone rods, etc.) provides many appealing features. First, sample extraction, clean-up, and concentration are conjugated in one step, enhancing reproducibility for sample preparation and throughput. Second, if operated in non-depletive mode, passive sampling allows insights of chemical activity because spontaneous processes such as partitioning will be driven by activity gradients until equilibrium is reached.110,111 Furthermore, the use of passive sampling devices provides a good balance between hydrophilic (mainly in freely dissolved form) and hydrophobic species (mainly associated with matrix components but usually with stronger affinity for polymeric phases),40 and is a promising technique for in situ104 and in vivo112 applications. The potential of wearable passive samplers to measure a large array of chemicals across individuals was recently reviewed.113 Their low-cost, noninvasive nature make them practical for large-scale deployment and integration with ecological momentary assessments enables the spatiotemporal variability of an individual’s chemical exposure to be captured. However, attention must be paid to potential artifacts with careful consideration to placement and sampling rate alteration, e.g. through obstruction such as coverage by clothing, personal care product use if in contact with skin, air velocity alterations for individual behaviors, etc.113
Sorbent choice is critical and most metabolomics applications have favored commercially available polydimethylsiloxane/carboxen/divinylbenzene (PDMS/Car/DVB) to provide broad coverage. Although currently limited to coatings made in lab, (HLB) particles have been shown to provide good performances in terms of sensitivity and extend coverage toward more polar analytes.100,114,115 For example, Vasiljevic et al. developed biocompatible SPME minitips coated with HLB particles and demonstrated their application for metabolomics and the targeted analysis of drugs of abuse,115 a combination polystyrene-divinylbenzene-weak anion exchange (PS-DVB-WAX)/HLB (50/50, w/w) fiber was demonstrated to cover a wide range of hydrophobicity (−7 < log P < 17) and molecular weight (100-1000 Da)116 and Gionfriddo et al. proposed a polytetrafluoroethylene amorphous fluoroplastics (PTFE AF)/HLB combination suitable for both GC and LC applications.114 Xu et al. developed a biocompatible and stable polystyrene–polydopamine–glutaraldehyde SPME coating for in vivo analysis on pharmaceuticals in fish muscle and demonstrated sensitivities from 26 up to 42 times larger than traditional PDMS fibers,117 evidencing the potential for application of novel sorbents for the in vivo sampling of environmental chemicals.118
The main pitfalls of passive sampling for non-target applications are selectivity (similarly to SPE or extraction by solvents), extraction of unbound (freely dissolved) molecules, and lengthy sampling times; though these can be tackled via addition of matrix modifiers (e.g. salts, surfactants) and high temperatures can speed up sampling rate,119–122 albeit with risk of increased degradation. When used for qualitative analysis or measurements of total concentrations, pushing passive sampling boundaries toward depletive mode by increasing the volume of the sampler (e.g. thickness of the SPME fiber) and or enhancing desorption from matrix (e.g. by using surfactants) to increase the amount of chemical extracted, can improve sensitivity.
Instrumental analysis
Advances in GC-HRMS analysis
Compared to LC-HRMS, GC-HRMS remains an underexploited tool in metabolomics and chemical exposomics, despite providing complementary compound coverage. GC separations can provide more reproducible retention than LC, with use of retention indices commonplace. Plus, the predominant mode of EI is largely standardized which enables highly reproducible fragmentation. The drawbacks are the limited range of compounds amenable to GC alongside the fact that EI is a “hard” (i.e. highly energetic) ionization mode and hinders annotation due to extensive in-source fragmentation. Furthermore, there is a substantial lack of open GC-HRMS spectral libraries123 and spectra collected in low resolution may be not fully comparable to data acquired in HR in the abundances of certain ions.124
Optimizing GC
Optimizing gas (and liquid) chromatography typically requires finding the best compromise between peak capacity/separation and sample throughput, selecting the best performing stationary phase and column characteristics (e.g. length, diameter, particle size), fitting the purposes of the work. An often overlooked approach to speedup the GC run and providing large sensitivity is the low pressure (so-called “vacuum-outlet”) GC. This consists in connecting a restrictor in the inlet to a short (10-15 m) wide (typically i.d. ≥ 0.5 mm) column as vacuum outlet for the MS.18,125 High vacuum reduces the viscosity of the gas mobile phase allowing for a higher flow rate and mass transfer. Compared to other fast-GC approaches like using short capillary columns, fast thermal gradients or high flow rates, this method is much more robust because of the use of large columns. The numerous benefits of low-pressure GC led some authors to recommend it as the standard approach for GC analysis,18 with promising applications to HRMS suspect/non-target screening.126 Possible drawbacks of this method regards increase of the column bleed, and potential narrowing of the peaks to durations incompatible with MS detector scan speed.126
Comprehensive two-dimensional GC (GC × GC) has been identified as a promising strategy for non-target analysis of environmental chemicals, providing greater peak capacity separations and often greater sensitivity.127 The theory128 and advances of this approach have been discussed for applications in, e.g. metabolomic,129 biomedical,130 and environmental131 research. Briefly, two GC columns are connected in series and multiple fractions/whole sample is sent from the first to the second column via a modulator. The increased commercial availability of GC × GC modulators looks set to push GC × GC towards routine use in the coming years.132 However, the reproducibility of GC × GC remains to be demonstrated for large-scale profiling applications, particularly studies that would necessitate intervening maintenance (e.g. column replacements).
GC-MS ionization sources
The high fragmentation and absence/low intensity of a molecular ion when using hard ionization (e.g. EI) poses challenges for processing and annotation workflows. Using lower ionization energies in EI has been proposed to reduce fragmentation.127 However, conflicting results have been reported in literature with this approach shown to be highly dependent upon source design.133 For example, significant improvements of molecular ion peak detection at 30 vs. 70 eV with a quadrupole-time of flight-mass spectrometer (Q-ToF-MS) were demonstrated,134 while just slight differences between 12 and 70 eV were observed via GC-Orbitrap.135 Another approach to enhance molecular ions is the use of Cold EI whereby a supersonic molecular beam interfaces the GC and MS and EI+ conducted on cold molecules.136 This was later pushed further toward soft ionization by reducing electron energy (from 70 eV to 18 eV) and lowering helium pressure to limit collisions via increased nozzle-skimmer distance, which effectively limited in source fragmentation and was successful to obtain mainly molecular ions.137 Cold EI has subsequently been linked to both LC and GC and shown favorable to ESI and conventional EI for identification,138 yet only tested at low resolution.
The use of “soft” (i.e. low energy) ionization such as chemical ionization (CI), field ionization (FI), photoionization (PI), or APCI is relatively widespread. Recently the coverage domain of EI and CI (positive and negative mode) was investigated on a commercial set of metabolites and shown to be complementary.139 Notably, of the 330 total compounds detected 81, 39, and 28 were unique for EI+, positive CI, and negative CI, respectively. However, sensitivity and coverage of soft ionization are on average reduced compared to EI+, and application usually additional rather than replacement for comprehensive profiling, decreasing throughput.127 Notably, electron capture negative ionization/negative CI is widely applied for highly sensitive targeted analysis of chemicals containing groups with high electronegativity, particularly halogens. Although overlooked in chemical exposomics (Table S4.1), recent applications have shown promise for non-target analysis.140–142
Advances in LC-HRMS analysis
LC coupled to HRMS enables separation and detection of a wide array of compounds in a diverse spectrum of physicochemical properties, providing maybe the best compromise between broad analytical coverage and simple sample treatment.124,143 However, results obtained in terms of analytical detection, sensitivity, and separation, largely depend on specific conditions used since different columns phases, modifiers, and chromatographic run length and temperatures can remarkably affect analytical outcomes. Advances in LC have been fundamental for the development of many broad range suspect/non-target methods. Notably, the introduction of ultra-high-pressure LC (UHPLC) with sub 2-micron columns enabled higher separation efficiencies,144 while core-shell/fused-core particles extended similar advantages to conventional high-pressure LC (HPLC).145 To capture the large diversity of chemicals, novel developments should be addressed toward the combination/testing of different stationary and mobiles phase chemistry, chromatographic conditions, and LC-MS interfaces.
Optimizing LC
Stationary phase
RP LC using C18 columns is typically favored because of the reliability and robustness, providing broad coverage, highly reproducibility retention times, and pH stability. Periat et al. tested 59 different columns for separation of 120 analytes which covered a wide range of hydrophobicity (−4.1 < log P < 14.76), using RPLC and hydrophilic interaction chromatography (HILIC), including mixed-mode columns and various pH conditions.146 Pentabromobenzyl (PBrBz) was recommended as a versatile broad-spectrum stationary phase while bridged ethylene hybrid (BEH) C8 at pH 9 and charged surface hybrid non-endcapped propylfluorophenyl columns were recommended for basic and acid compounds, respectively. Despite orthogonality, the additional benefits of complementing RP separations with HILIC were deemed to be low, offering just a 1% increase in unique chemicals showing acceptable peak shape beyond that possible for combining different RPs.146 Conversely, Contrepois et al. argued high complementarity by showing that fifty percent of the standard compound that showed unsatisfactory separation response via HILIC were acceptably detected through RPLC.147 Similarly, Zhang et al. compared an RP, two HILIC, and two aqueous normal phase separations for 176 metabolite standards, and demonstrated that complementary use of the two HILIC methods attained the greatest coverage.148
Further diversification of reversed-phase column choice also offers a rapid way to enhance coverage. For example, a combination of C8 and T3 (C18 modified for improved retention of polar groups) columns quadrupled the number of non-polar to semi-polar standards detectable compared to use of a single conventional C18 column.149 Also, the application of C30 column showed better performance for separation for 49 lipid standards150 and alternate selectivity for polar and non-polar analytes,151 yet are rarely used in comparison to traditional C18 columns. In addition, the use of porous graphitized carbon has been suggested as a RP alternative to HILIC separation, though considerations are needed to maintain retention stability152 and optimal choice between each dependent on analytes investigated and sample matrix.153
The benefits and drawbacks of coupling of column with different phase chemistry and separation mechanism in series, in parallel or offline have recently been discussed.154 Notably, many metabolomic studies have applied complementary RPLC and HILIC to simultaneously screen hydrophilic and hydrophobic chemicals,147,155–157 and the approach could be similarly applied in chemical exposomics.
Column dimensions (scale)
The miniaturization of LC-MS has recently been extensively reviewed158–160 and was highlighted by David et al. as a core development need to improve characterization of the human internal chemical exposome.24 Miniaturization can increase sensitivity via enhanced ionization efficiency/reduced ion suppression,161,162 reduced sample dilution, and greater resolution i.e. sharper peaks.163,164 Furthermore, miniaturization reduces solvent consumption165 and has been demonstrated to have reproducible performance with suitability for large-scale studies, including metabolomics applications.166
Microscale columns (i.d.: 0.5-1 mm162) provided good performances compared to narrow bore scale (2 mm162) in metabolomic studies, by increasing sensitivity, signal to noise ratio (S/N), and number of features identified (Table 2), although some system updates (e.g. reduction of the tubing and ESI capillary diameters) can be necessary.164 Moreover, Gray et al. reported excellent stability of microbore LC over time with no significant changes of peak shape, intensity, and backpressure after 1000 consecutive injections of human urine.164 Similarly, experiment with capillary columns (i.d.: 0.1-0.5mm)162 provided much better sensitivity than narrow bore scale.167,168 Nanoscale LC-ESI systems work with columns of 0.01-0.1 mm i.d.,162 flow rates of 10-1000 nL/min and ESI capillary with i.d. of 10-50 μm166 and offer a better sensitivity than conventional narrow bore and microscale due to droplets 100-1000 fold smaller and reduced sample dilution.169 For example, a 2- to 2000-fold improvement in detection limits has been demonstrated when reducing from a 1 mm to 0.1 mm column (Table 2).170
Performance comparisons of miniaturized LC and conventional narrow bore scale.
| Internal diameter (mm) | Flow rate (µL/min) | Matrix | Results | Reference |
|---|---|---|---|---|
| 1 vs. 2.1 | 136 vs. 500 | Rat urine | Increase of S/N ratio (20% for a feature), reduction of the sample injected (from 10 to 2 µL) | Lenz et al.161 |
| 1 vs. 2.1 | 140 vs. 600 | Rat plasma | Increase in S/N (up to 3.6-fold across 5 pharmaceuticals), decrease of the solvent consumed (∼ 4-fold) | Rainville et al.165 |
| 1 vs. 2.1 | 140 vs. 600 | Human urine | Increase in peak area, peak height, and S/N of 2- to 3-fold for metabolites (7 analyzed in ESI+ and 4 in ESI−) and ∼10% more features detected | Gray et al.164 |
| 0.32 vs. 2.1 | 10 vs. 600 | Rat urine | Increase in sensitivity of ∼100-fold and doubled ion detection | Granger et al.167 |
| 0.2 vs. 2.1 | 6 vs. 700 | Human plasma | Increase in sensitivity (peak areas) from 6- to 49-fold among 11 drugs | Zhang et al.168 |
| 0.1 vs. 1 | 0.7 vs. 200 | 50/50 MeOH/water | Decrease of the instrumental limits of detection from a factor 2 to a factor of 2000, for 78 xenometabolites | Chetwynd et al.170 |
However, compared to microscale and capillary separations major drawbacks of nanoscale LC are lowered robustness (e.g. column connections, tubing) increasing maintenance, slow conditioning and equilibration times,171 and the need for more careful sample preparation and purification required to avoid the clogging of the system.54,166 Each of these drawbacks reduces throughput and will need to be overcome for application to population-based chemical exposomics. To this end, applications of nano-UHPLC separations (i.e. using small diameter, small particle size columns) are increasing.171 Recently commercialized micropillar array columns with enhanced robustness and reproducibility are showing promise in proteomics172 and the coupling of SPME with nano-LC represents a promising approach to introduce a highly purified sample115,173 without the need of extensive clean-up steps.
Mobile phase modifiers
Most exposome studies used formic acid (typically 0.1%, with or without salt addition, e.g. ammonium acetate/formate) in water/methanol or water/acetonitrile gradients (Tables 1 and S3.1). However, promising results have been achieved with other modifiers. For example, Yanes et al. compared ammonium fluoride (NH4F; 1 mM) against ammonium acetate (5 mM) and formic acid (FA; 0.1%) on a conventional C18 column in ESI− and observed an average increase of 5.7-fold of the signal intensity for 36 standards.14 Ammonium fluoride has also been used for enhanced detection of emerging contaminants up to 5-fold174 and xenoestrogens by RPLC, claimed favorable for both positive and negative ionization.156 Similar results were obtained for HILIC, where NH4F (2 mM) provided better ionization efficiency and signal-to-noise ratios than ammonium acetate (10 mM) for 34 standards via ESI−.176 However, attention must be paid when handling NH4F solutions for potential hydrogen fluoride degassing.175 Furthermore, studies on stability of this modifier over time should be performed since it may be microbiologically less stable than acidified solutions. Zhang et al. instead recommended acetic acid (1 mM) for improved detection of 26 metabolite standards when compared to both NH4F and FA across a range of additive concentrations.177 From a test of 17 different modifier conditions in ESI+ and 23 in ESI−, the observed compound-specific responses led to recommendation of using a combination of different modifiers for various ionization modes and analytes.178 Furthermore, monitoring adduct mass and intensity shifts from use of isotopically enriched modifiers has recently been demonstrated to aid identification of potential metabolite features,179 and could equally be applied to chemical exposomics.
LC temperatures
The use of high-temperature (HT) LC to increase separation efficiency and enable faster separations is a potential approach to increase throughput.180 The decreased viscosity of the mobile phase can facilitate use of unconventional solvents (e.g. ethanol, isopropanol) to increase coverage, enable a reduction of the use of organic modifiers for elution and increase column lifetime by enhanced removal of, e.g. phospholipid matrix interferences. The main pitfalls are the limited availability of thermostable columns, potential loss of thermolabile compounds, and the need for dedicated instrumentation, e.g. pre-heating apparatus for the mobile phase, pressure regulation interface with MS.181,182 Though maximizing temperature is recommended for separation efficiency,181 the effect of mobile phase temperature on MS detection sensitivity was shown to be compound specific and gains in analyte signal intensity can be offset by increasing noise.183 Despite this, applications utilizing HT-UHPLC-MS have demonstrated the benefits of increased ion detection and greater separation compared to conventional RPLC, for detection of both endogenous and exogenous compounds.184,185 This approach has been successful in reducing run time186,187 providing good peak capacity in rat and human urine metabolomic analysis, both by using conventional isotherm (90°C) with solvent gradient184 and thermal gradient (up to 180°C).188 These studies showed that reproducible HT-UPLC-MS is achievable for complex matrices (e.g. blood plasma), though the authors note HT LC still requires further development.
LC-MS ionization sources
Novel interfaces could extend LC-MS coverage and sensitivity. The use of multiple polarity and ionization modes has been evidenced to enhance coverage for metabolomics189 and rapid polarity switching and dual/multimode sources common.190 Rapid polarity switching can be less robust than separate acquisition (e.g. through disturbed spray) and increases cycle times limiting scans per peak; though dual emitters help tackle each problem.191 The main limitation is that the use of a single mobile phase can compromise coverage.192 Multimode ionization tends to show a reduction in sensitivity.193 Alternatively, rapid switching between ESI and APCI sources was recently demonstrated without ion abundance loss and is a promising option for further development.194 Novel ionization processes have been recently reviewed.195 In particular, the electrospray ionization inlet (ESII) shows promise, offering increased ion abundance and lower background signal when compared to ESI, able to provide similar advantages as nano-ESI at microscale flowrates.196
Finally, research advances coupling LC with EI ionization are rapidly progressing toward commercial viability.197–199 Combining the advantages of LC separations with EI ionization could significantly enhance coverage and provides more reproducible fragmentation though, at present, sensitivity requires improvement to be competitive with ESI. Optimizing source parameters (e.g. by using design of experiment, DoE) can improve analyte detection.15,200 For example, high in-source fragmentation was recently demonstrated to increase the proportion of annotated coverage.201 Notably, in silico method development enables rapid testing and optimization of acquisition parameters202,203 while technological advances have seen the emergence of information-dependent acquisition modes to enable automated real-time parameter optimizations during analysis.204–206
An assessment of costs, benefits, and potential for chemical exposomics
Strategies reviewed in the previous section were assessed on the basis of their benefits and costs and a division proposed sorting out strategies with (i) low costs and high benefits; (2) low costs and low benefits; (3) high costs and high benefits; (4) high costs and low benefits (Table 3). Benefits considered were in terms of sensitivity (enhanced recovery/sample enrichment or instrumental response); physicochemical range coverage; reproducibility; sample throughput; practicality (strategies easy to apply, potential for automation/reducing manual operations, etc.). Costs accounted for were capital expenditure (dividing minor and major investment for capital equipment); installation (ready to use items vs. need for installations/specialist support); operational costs associated with maintenance (long-term financial costs to keep functionality); consumables (financial cost plus environmental burden), and commercial availability (niche/in-house applications were considered demanding high efforts). Access to conventional LC and GC-HRMS instrumentation was considered a prerequisite.
Assessment of analytical strategies for chemical exposomics
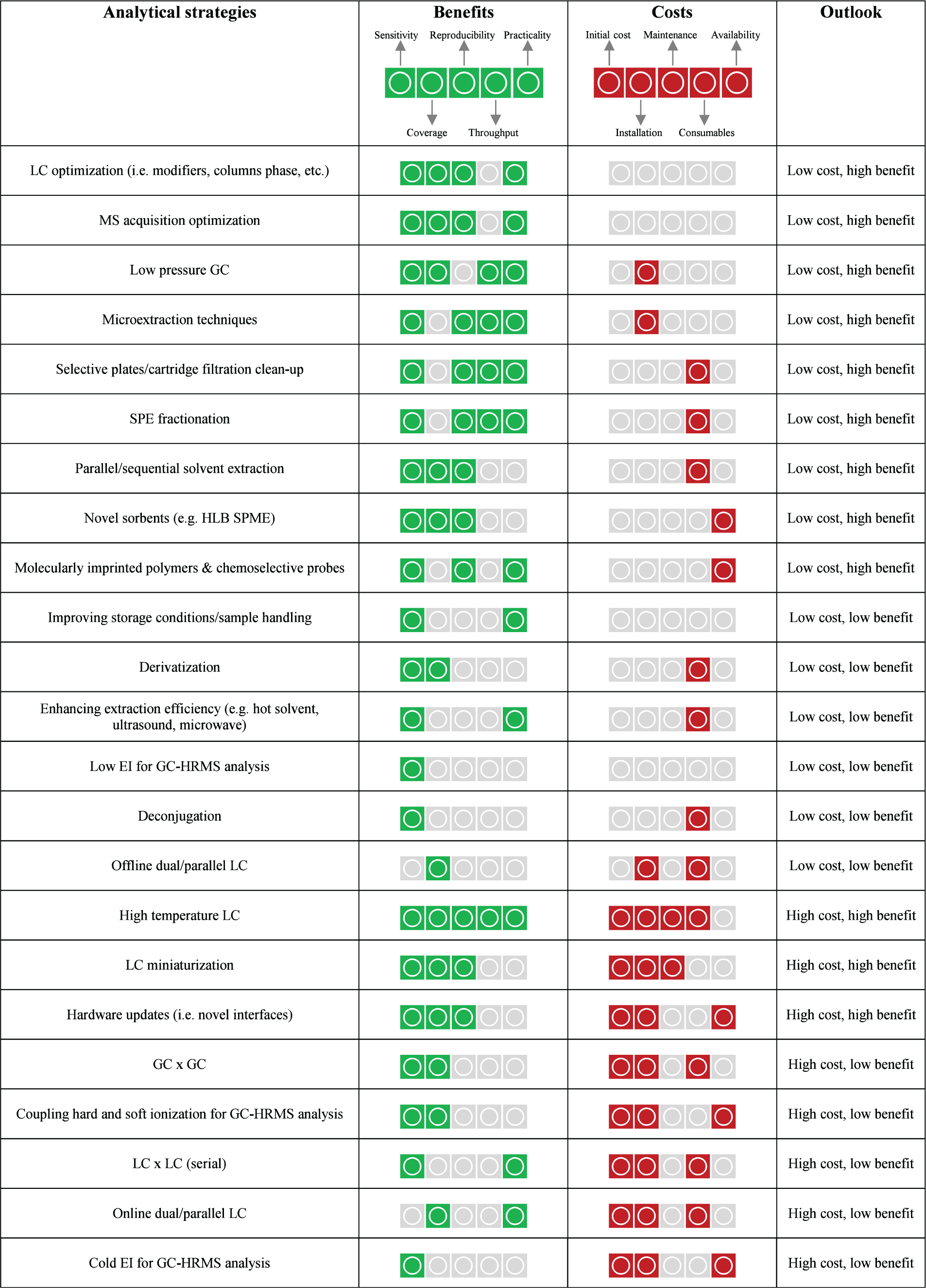 |
Colored criteria are fulfilled.
Ideally, strategies with high benefit and limited costs/efforts should be readily tested and include alternative sample enrichment methods, more refined purification methods, chromatographic optimizations (Table 3). Promising results in enhancing coverage and detection of low abundant compounds have been obtained with sequential extractions and fractionations, microextraction techniques, and chemoselective probes which encourage their application for non-target analysis of environmental contaminants.
Some of these approaches have been widely applied in target studies while still underexploited for non-targeted analysis (e.g. SPME). However, commercially available materials (e.g. HLB SPME, novel imprinted polymers/probes) should be developed to simplify analysis and reduce manual operations. Fractionation methods, with different solvents or sorbent extraction, are included in recently developed workflows,59,67 and offer the benefit of differential enrichment of fractions and/or analysis via multiple platforms. Whenever possible, preparation methods should merge the steps needed for sample purification. For example, interferences that ionize in multiple modes and/or have detrimental effects on columns and signal suppression (e.g. lipids) should be removed prior to fractionation. Similarly, enzymatic/chemical treatments (e.g. protease/lipase treatment/derivatization) could be implemented prior to extraction, and may enable more selective clean-up.
Optimized chromatographic settings from other LC-HRMS-based omics (e.g. ammonium fluoride vs. formic acid; PBrBz vs. C18-based separation) should be more widely tested as they show potential to be largely beneficial with virtually no costs. Target studies support the potential of these strategies (e.g. utilization of ammonium fluoride as modifier).174,175
Low-pressure GC has shown improved sensitivity and potential for routine implementation with minor hardware updates. This is a largely overlooked approach in chemical exposomics that should be prioritized for implementation, especially considering the possibility of loading large sample volumes that could be a valuable asset for low abundant chemicals. Remarkable improvements in instrumental sensitivity can be obtained just optimizing MS acquisition parameters with proper experimental design. Although this is commonly performed in-house, harmonized guidelines on how to do this when a broad range of chemicals is investigated are lacking.
The combination of GC and LC separation via workflows that separate polar and non-polar compounds prior to analysis would capitalize on the advantages of each platform. Compared to minimal sample preparation, this would allow to introduce cleaner extracts in the system, since potential matrix interferences would partition as well, and thus both coverage and sensitivity can be enhanced.
If limited resources are available, strategies with low costs and some benefits could be implemented (Table 3). These include mainly minor laboratory tips for sample handling/storage conditions and strategies to improve recovery (e.g. hot solvent and ultrasound extractions) and/or to obtain larger coverage (e.g. derivatization). Limited studies, for example, are available on chemical stability over storage and potential of deconjugation76 and derivatization92 for suspect and untargeted workflows.
Reducing pitfalls of very promising strategies currently with elevated costs (Table 3) should be the goal of next research developments. These include advanced hardware developments with innovative interfaces that allow for better ionization and instrumental sensitivity, though requiring development to become commercially available. For example, HT LC appears a promising strategy to enhance mass transfer and allow for high throughput analysis, yet is currently is limited by high costs and maintenance. LC miniaturization needs to be coupled with advanced sample cleaning that can limit throughput. These strategies are largely unexplored in chemical exposomics.
Outlook and conclusions
A survey of the analytical conditions used in chemical exposomics underlined the need to diversify current methods and approaches to advance toward more comprehensive measures of chemical exposures. To enhance coverage and detection of low abundant environmental chemicals, a proficient use of knowledge and experience inherited from related fields was previously advocated. Therefore, the analytical approaches used in HRMS-based metabolomics and proteomics were reviewed, along with newly emerging wide-scope multiresidue target analysis methods, to identify potential strategies suitable for chemical exposomics. A pragmatic assessment of the costs and potential benefits of implementing the strategies in chemical exposomics was performed to prioritize those with most promise (Table 3). We hope this provides a practical aid for research groups developing chemicals exposomics and guides investments to address next research.
Beside analytical challenges, data processing constraints hinder the development of a comprehensive characterization of the chemical exposome and represent a bottleneck for untargeted workflows.24,27 Computational tools must cope with low intensities features at noise level, scarcity of the signals across samples, and large variation of the chemical response, due for example to matrix interferences, variations in dilution factors, and recovery issues; with consequent challenges for the statistical analysis of the data. The profiling of endogenous compounds has attained comparably high compound coverage in terms of detection and annotation than for the profiling of exogenous chemicals in biospecimen. For example, measurement of ∼100s-1000s of endogenous metabolites per human plasma sample is possible by LC-HRMS, compared to just 10s-100s commonly found exogenous chemicals via the same method/platform.207 The divide is largely a result of the average lower internal abundance of exogenous chemicals, coupled with greater inter- and intra-individual variability, and limitations in the detection and annotation/identification of low abundant features during data processing. It should be considered that the strategies proposed here should also be supported with modification and development of the data processing workflows, especially for the aggregation and/or integration of multiple diverse datasets acquired via complementary workflows.
Furthermore, harmonized standard procedures, quality assurance (QA)/ quality control (QC) protocols, and minimum reporting standards should be implemented for chemical exposomics, similar to efforts in, e.g. metabolomics42,208,209 to assess overall quality of the data produced and comparability across laboratories. Steps toward this direction have been recently proposed.210,211 Ideally, the recommended guidelines should be as streamlined and simple as possible,53,212 to avoid encountering a lack of uptake scenario, commonly exampled in metabolomics.213,214
To conclude, we wish that this manuscript encourages further exchanges and discussions across experts from different fields. In our vision, sharing knowledge, tools, and analytical hints across fields is fundamental to push forward the limits of the current approaches and integrate findings. Integrating experiences from related fields is a critical step to avoid reinventing the wheel in the emerging field of chemical exposomics. A plethora of approaches and novel strategies must be developed, tested, and integrated to push toward more comprehensive measures of chemical exposures and there is no time to waste.
Supplementary material
Supplementary material is available at Exposome online.
Funding
C.M.V., E.J.P., and J.K. thank to Research Infrastructure RECETOX RI (No. LM2018121) and the project CETOCOEN EXCELLENCE (No. CZ.02.1.01/0.0/0.0/17_043/0009632) financed by the Ministry of Education, Youth and Sports for supportive background. E.J.P. acknowledges support from Operational Programme Research, Development and Innovation - project Postdoc@MUNI (No. CZ.02.2.69/0.0/0.0/16_027/0008360) and MSCAfellow4@MUNI (No. CZ.02.2.69/0.0/0.0/20_079/0017045). This work has received funding from the European Union’s Horizon 2020 research and innovation programme under grant agreement No 874627. This publication reflects only the authors's view and the European Commission is not responsible for any use that may be made of the information it contains. R.B. acknowledges support from INSERM and Université de Paris to Unit 1124.
Conflict of interest statement
G.W.M. receives royalties for his books The Exposome: A Primer and The Exposome: A New Paradigm for the Environment and Health. He has no other conflicts of interest. The other authors declare they have no actual or potential competing financial interests.
Authors’ contributions
All authors contributed to the work presented in this manuscript, provided final approval and are accountable.
Data availability
The authors confirm that the data supporting the findings of this study are available within the article and its supplementary materials.
References
1SoltowQA, StrobelFH, MansfieldKG, WachtmanL, ParkY, JonesDP. High-performance metabolic profiling with dual chromatography-Fourier-transform mass spectrometry (DC-FTMS) for study of the exposome. Metabolomics 2013; 9(1 Suppl):S132–S143. doi:10.1007/s11306-011-0332-1 ↩
2WarthB, SpanglerS, FangM, et al Exposome-scale investigations guided by global metabolomics, pathway analysis, and cognitive computing. Anal Chem. 2017; 89(21):11505–11513. doi:10.1021/acs.analchem.7b02759 ↩
3BonvallotN, DavidA, ChalmelF, et al Metabolomics as a powerful tool to decipher the biological effects of environmental contaminants in humans. Curr Opinion Toxicol. 2018; 8:48–56. doi:10.1016/j.cotox.2017.12.007 ↩
4RagerJE, StrynarMJ, LiangS, et al Linking high resolution mass spectrometry data with exposure and toxicity forecasts to advance high-throughput environmental monitoring. Environ Int. 2016; 88:269–280. doi:10.1016/j.envint.2015.12.008 ↩
5AthersuchTJ, KeunHC. Metabolic profiling in human exposome studies. MUTAGE 2015; 30(6):755–762 [published online ahead of print]. doi:10.1093/mutage/gev060 ↩
6EscherBI, HackermüllerJ, PolteT, et al From the exposome to mechanistic understanding of chemical-induced adverse effects. Environ Int. 2017; 99:97–106. doi:10.1016/j.envint.2016.11.029
7BocatoMZ, Bianchi XimenezJP, HoffmannC, BarbosaF. An overview of the current progress, challenges, and prospects of human biomonitoring and exposome studies. J Toxicol Environ Health B Crit Rev. 2019; 22(5-6):131–156. doi:10.1080/10937404.2019.1661588
8KraussM, SingerH, HollenderJ. LC–high resolution MS in environmental analysis: from target screening to the identification of unknowns. Anal Bioanal Chem. 2010; 397(3):943–951. doi:10.1007/s00216-010-3608-9 ↩
9SchymanskiEL, SingerHP, SlobodnikJ, et al Non-target screening with high-resolution mass spectrometry: critical review using a collaborative trial on water analysis. Anal Bioanal Chem. 2015; 407(21):6237–6255. doi:10.1007/s00216-015-8681-7
10DiasD, JonesO, BealeD, et al Current and future perspectives on the structural identification of small molecules in biological systems. Metabolites 2016; 6(4):46.doi:10.3390/metabo6040046
11HollenderJ, SchymanskiEL, SingerHP, FergusonPL. Nontarget Screening with High Resolution Mass Spectrometry in the Environment: Ready to Go? Environ Sci Technol. 2017 Oct 17; 51(20):11505-11512. doi: 10.1021/acs.est.7b02184 ↩
12WantEJ, NordströmA, MoritaH, SiuzdakG. From exogenous to endogenous: the inevitable imprint of mass spectrometry in metabolomics. J Proteome Res. 2007 Feb; 6(2):459-68. doi: 10.1021/pr060505+
13HuM, KraussM, BrackW, SchulzeT. Optimization of LC-Orbitrap-HRMS acquisition and MZmine 2 data processing for nontarget screening of environmental samples using design of experiments. Anal Bioanal Chem. 2016; 408(28):7905–7915. doi:10.1007/s00216-016-9919-8 ↩
14YanesO, TautenhahnR, PattiGJ, SiuzdakG. Expanding coverage of the metabolome for global metabolite profiling. Anal Chem. 2011; 83(6):2152–2161. doi:10.1021/ac102981k ↩
15KnolhoffAM, KneaplerCN, CroleyTR. Optimized chemical coverage and data quality for non-targeted screening applications using liquid chromatography/high-resolution mass spectrometry. Anal Chim Acta. 2019; 1066:93–101. doi:10.1016/j.aca.2019.03.032 ↩
16SinghRR, ChaoA, PhillipsKA, et al Expanded coverage of non-targeted LC-HRMS using atmospheric pressure chemical ionization: a case study with ENTACT mixtures. Anal Bioanal Chem. 2020; 412(20):4931–4939. doi:10.1007/s00216-020-02716-3
17SouthamAD, LangeA, Al-SalhiR, HillEM, TylerCR, ViantMR. Distinguishing between the metabolome and xenobiotic exposome in environmental field samples analysed by direct-infusion mass spectrometry based metabolomics and lipidomics. Metabolomics. 2014; 10(6):1050–1058. doi:10.1007/s11306-014-0693-3 ↩
18SapozhnikovaY, LehotaySJ. Review of recent developments and applications in low-pressure (vacuum outlet) gas chromatography. Anal Chim Acta. 2015; 899:13–22. doi:10.1016/j.aca.2015.10.003 ↩
19SteinerD, SulyokM, MalachováA, MuellerA, KrskaR. Realizing the simultaneous liquid chromatography-tandem mass spectrometry based quantification of >1200 biotoxins, pesticides and veterinary drugs in complex feed. J Chromatogr A. 2020; 1629:461502.doi:10.1016/j.chroma.2020.461502
20MonteiroSH, LehotaySJ, SapozhnikovaY, NingaE, LightfieldAR. High-throughput mega-method for the analysis of pesticides, veterinary drugs, and environmental contaminants by ultra-high-performance liquid chromatography−tandem mass spectrometry and robotic mini-solid-phase extraction cleanup + low-pressure gas chromatography−tandem mass spectrometry, part 1: beef. J Agric Food Chem. 2021; 69(4):1159–1168. doi:10.1021/acs.jafc.0c00710
21NingaE, SapozhnikovaY, LehotaySJ, LightfieldAR, MonteiroSH. High-throughput mega-method for the analysis of pesticides, veterinary drugs, and environmental contaminants by ultra-high-performance liquid chromatography–tandem mass spectrometry and robotic mini-solid-phase extraction cleanup + low-pressure gas chromatography–tandem mass spectrometry, part 2: catfish. J Agric Food Chem. 2021; 69(4):1169–1174. doi:10.1021/acs.jafc.0c00995
22MetzTO, BakerES, SchymanskiEL, et al Integrating ion mobility spectrometry into mass spectrometry-based exposome measurements: what can it add and how far can it go? Bioanalysis 2017; 9(1):81–98. doi:10.4155/bio-2016-0244 ↩
23WestC. Current trends in supercritical fluid chromatography. Anal Bioanal Chem. 2018; 410(25):6441–6457. doi:10.1007/s00216-018-1267-4 ↩
24DavidA, ChakerJ, PriceEJ, BessonneauV, ChetwyndAJ, VitaleCM, KlánováJ, WalkerDI, AntignacJP, BaroukiR, MillerGW. Towards a comprehensive characterisation of the human internal chemical exposome: Challenges and perspectives. Environ Int. 2021 Nov; 156:106630. doi: 10.1016/j.envint.2021.106630 ↩
25PriceEJ., VitaleCM., MillerGW., DavidA, Barouki RAudouze, WalkerDI., AntignacJ-P, CoumoulX, BessonneauV, KlánováJ. (2021). Merging the exposome in an integrated framework for "omic" sciences (2.0.1). Zenodo. https://doi.org/10.5281/zenodo.5579746https://doi.org/10.5281/zenodo.5579746 ↩
26SchymanskiEL, SingerHP, LongréeP, et al Strategies to characterize polar organic contamination in wastewater: exploring the capability of high resolution mass spectrometry. Environ Sci Technol. 2014; 48(3):1811–1818. doi:10.1021/es4044374 ↩
27PourchetM, DebrauwerL, KlanovaJ, PriceEJ, CovaciA, Caballero-CaseroN, OberacherH, LamoreeM, DamontA, FenailleF, VlaanderenJ, MeijerJ, KraussM, SarigiannisD, BaroukiR, Le BizecB, AntignacJP. Suspect and non-targeted screening of chemicals of emerging concern for human biomonitoring, environmental health studies and support to risk assessment: From promises to challenges and harmonisation issues. Environ Int. 2020 Jun; 139:105545. doi: 10.1016/j.envint.2020.105545 ↩
28AndraSS, AustinC, PatelD, DoliosG, AwawdaM, AroraM. Trends in the application of high-resolution mass spectrometry for human biomonitoring: an analytical primer to studying the environmental chemical space of the human exposome. Environ Int. 2017; 100:32–61. doi:10.1016/j.envint.2016.11.026 ↩
29MilmanBL, ZhurkovichIK. The chemical space for non-target analysis. Trends Anal Chem. 2017; 97:179–187. doi:10.1016/j.trac.2017.09.013
30RappaportSM, BarupalDK, WishartD, VineisP, ScalbertA. The blood exposome and its role in discovering causes of disease. Environ Health Perspect. 2014; 122(8):769–774. doi:10.1289/ehp.1308015 ↩
31OrešičM, McGlincheyA, WheelockCE, HyötyläinenT. Metabolic Signatures of the Exposome-Quantifying the Impact of Exposure to Environmental Chemicals on Human Health. Metabolites. 2020 Nov 10; 10(11):454. doi: 10.3390/metabo10110454
32HyötyläinenT. Analytical challenges in human exposome analysis with focus on environmental analysis combined with metabolomics. J Sep Sci. 2021; 44(8):1769–1787. doi:10.1002/jssc.202001263
33StevensVL, HooverE, WangY, ZanettiKA. Pre-Analytical Factors that Affect Metabolite Stability in Human Urine, Plasma, and Serum: A Review. Metabolites. 2019 Jul 25; 9(8):156. doi: 10.3390/metabo9080156 ↩
34BiH, GuoZ, XiaoJ, HuiyingL, LulinM, LixiangX. The key points in the pre-analytical procedures of blood and urine samples in metabolomics studies [published online ahead of print]. Metabolomics. 2020; 16(6):68. doi:10.1007/s11306-020-01666-2 ↩
35González-DomínguezR, González-DomínguezÁ, SayagoA, Fernández-RecamalesÁ. Recommendations and Best Practices for Standardizing the Pre-Analytical Processing of Blood and Urine Samples in Metabolomics. Metabolites 2020; 10(6) http://doi.org/10.3390/metabo10060229 32503183
36PerryJN, JasimA, HojatA, YongWH. Procurement, storage, and use of blood in biobanks. In: YongWH, ed. Biobanking. Vol 1897. Methods in Molecular Biology. Springer New York; 2019:89–97. doi:10.1007/978-1-4939-8935-5_9 ↩
37KirwanJA, BrennanL, BroadhurstD, FiehnO, CascanteM, DunnWB, SchmidtMA, VelagapudiV. Preanalytical Processing and Biobanking Procedures of Biological Samples for Metabolomics Research: A White Paper, Community Perspective (for "Precision Medicine and Pharmacometabolomics Task Group"-The Metabolomics Society Initiative). Clin Chem. 2018 Aug; 64(8):1158-1182. doi: 10.1373/clinchem.2018.287045 ↩
38DennisKK, MarderE, BalshawDM, et al Biomonitoring in the era of the exposome. Environ Health Perspect. 2017; 125(4):502–510. doi:10.1289/EHP474 ↩
39HebelsDGAJ, GeorgiadisP, KeunHC, et al, EnviroGenomarkers Project Consortium. Performance in omics analyses of blood samples in long-term storage: opportunities for the exploitation of existing biobanks in environmental health research. Environ Health Perspect. 2013; 121(4):480–487. doi:10.1289/ehp.1205657 ↩
40Reyes-GarcésN, GionfriddoE. Recent developments and applications of solid phase microextraction as a sample preparation approach for mass-spectrometry-based metabolomics and lipidomics. Trends in Anal Chem. 2019; 113:172–181. doi:10.1016/j.trac.2019.01.009 ↩
41BerendsenBJA, ElbersIJW, StolkerAAM. Determination of the stability of antibiotics in matrix and reference solutions using a straightforward procedure applying mass spectrometric detection [published online ahead of print October 11, 2011]. Food Addit Contam Part A Chem Anal Control Expo Risk Assess. 2011; 28(12):1657–1610. doi:10.1080/19440049.2011.604045 ↩
42DudzikD, Barbas-BernardosC, GarcíaA, BarbasC. Quality assurance procedures for mass spectrometry untargeted metabolomics. A review. J Pharm Biomed Anal. 2018; 147:149–173. doi:10.1016/j.jpba.2017.07.044 ↩
43YuM, PetrickL. Untargeted high-resolution paired mass distance data mining for retrieving general chemical relationships. Commun Chem. 2020; 3(1):157. doi: 10.1038/s42004-020-00403-z ↩
44RaymondJW, RogersTN, ShonnardDR, KlineAAA. review of structure-based biodegradation estimation methods. J Hazard Mater. 2001; 84(2-3):189–215. doi:10.1016/S0304-3894(01)00207-2 ↩
45FennerK, ScrepantiC, RenoldP, RouchdiM, VoglerB, RichS. Comparison of small molecule biotransformation half-lives between activated sludge and soil: opportunities for read-across? Environ Sci Technol. 2020; 54(6):3148–3158. doi:10.1021/acs.est.9b05104 ↩
46KisandK, KernaI, KummJ, JonssonH, TammA. Impact of cryopreservation on serum concentration of matrix metalloproteinases (MMP)-7, TIMP-1, vascular growth factors (VEGF) and VEGF-R2 in Biobank samples. Clin Chem Lab Med. 2011; 49(2):229–235. doi:10.1515/CCLM.2011.049 ↩
47WuY, LiL. Sample normalization methods in quantitative metabolomics. J Chromatogr A. 2016; 1430:80–95. doi:10.1016/j.chroma.2015.12.007 ↩
48MisraBB. Data normalization strategies in metabolomics: current challenges, approaches, and tools. Eur J Mass Spectrom (Chichester). 2020; 26(3):165–174. doi:10.1177/1469066720918446 ↩
49WarrackBM, HnatyshynS, OttK-H, et al Normalization strategies for metabonomic analysis of urine samples. J Chromatogr B Analyt Technol Biomed Life Sci. 2009; 877(5-6):547–552. doi:10.1016/j.jchromb.2009.01.007 ↩
50MalmL, PalmE, SouihiA, PlassmannM, LiigandJ, KruveA. Guide to Semi-Quantitative Non-Targeted Screening Using LC/ESI/HRMS. Molecules. 2021 Jun 9; 26(12):3524. doi: 10.3390/molecules26123524 ↩
51LiigandP, LiigandJ, KaupmeesK, KruveA. 30 Years of research on ESI/MS response: Trends, contradictions and applications. Anal Chim Acta. 2021 Apr 1; 1152:238117. doi: 10.1016/j.aca.2020.11.049 ↩
52BarupalDK, BaygiSF, WrightRO, AroraM. Data Processing Thresholds for Abundance and Sparsity and Missed Biological Insights in an Untargeted Chemical Analysis of Blood Specimens for Exposomics. Front Public Health. 2021 Jun 10; 9:653599. doi: 10.3389/fpubh.2021.653599 ↩
53AlseekhS, AharoniA, BrotmanY, el al. Mass spectrometry-based metabolomics: a guide for annotation, quantification and best reporting practices. Nat Methods. 2021 Jul; 18(7):747-756. doi: 10.1038/s41592-021-01197-1 ↩
54DavidA, Abdul-SadaA, LangeA, TylerCR, HillEM. A new approach for plasma (xeno)metabolomics based on solid-phase extraction and nanoflow liquid chromatography-nanoelectrospray ionisation mass spectrometry. J Chromatogr A. 2014; 1365:72–85. doi:10.1016/j.chroma.2014.09.001 ↩
55PourchetM, NarduzziL, JeanA, et al Non-targeted screening methodology to characterise human internal chemical exposure: Application to halogenated compounds in human milk. Talanta. 2021 Apr 1; 225:121979. doi: 10.1016/j.talanta.2020.121979 ↩
56TeoCC, ChongWPK, HoYS. Development and application of microwave-assisted extraction technique in biological sample preparation for small molecule analysis. Metabolomics 2013; 9(5):1109–1128. doi:10.1007/s11306-013-0528-7 ↩
57Luque de CastroMD, Delgado-PovedanoMM. Ultrasound: a subexploited tool for sample preparation in metabolomics. Anal Chim Acta. 2014; 806:74–84. doi:10.1016/j.aca.2013.10.053 ↩
58Ferreiro-VeraC, Priego-CapoteF, Luque de CastroMD. Comparison of sample preparation approaches for phospholipids profiling in human serum by liquid chromatography–tandem mass spectrometry. J Chromatogr A. 2012; 1240:21–28. doi:10.1016/j.chroma.2012.03.074 ↩
59YangY, CruickshankC, ArmstrongM, MahaffeyS, ReisdorphR, ReisdorphN. New sample preparation approach for mass spectrometry-based profiling of plasma results in improved coverage of metabolome. J Chromatogr A. 2013; 1300:217–226. doi:10.1016/j.chroma.2013.04.030 ↩
60FaulandA, TrötzmüllerM, EberlA, et al An improved SPE method for fractionation and identification of phospholipids: sample preparation. J Sep Sci. 2013; 36(4):744–751. doi:10.1002/jssc.201200708 ↩
61CajkaT, FiehnO. Comprehensive analysis of lipids in biological systems by liquid chromatography-mass spectrometry. Trends Anal Chem. 2014; 61:192–206. doi:10.1016/j.trac.2014.04.017 ↩
62SchemethD, NielsenNJ, AnderssonJT, ChristensenJH. A tiered analytical approach for target, non-target and suspect screening analysis of polar transformation products of polycyclic aromatic compounds. Chemosphere 2019; 235:175–184. doi:10.1016/j.chemosphere.2019.06.149 ↩
63KadokamiK, UenoD. Comprehensive target analysis for 484 organic micropollutants in environmental waters by the combination of tandem solid-phase extraction and quadrupole time-of-flight mass spectrometry with sequential window acquisition of all theoretical fragment-ion spectra acquisition. Anal Chem. 2019; 91(12):7749–7755. doi:10.1021/acs.analchem.9b01141 ↩
64MichopoulosF, EdgeA, HuiY-T, LiddicoatT, TheodoridisG, WilsonI. Extraction methods for the removal of phospholipids and other endogenous material from a biological fluid. Bioanalysis 2011; 3(24):2747–2755. doi:10.4155/bio.11.283 ↩
65BaduelC, MuellerJF, TsaiH, Gomez RamosMJ. Development of sample extraction and clean-up strategies for target and non-target analysis of environmental contaminants in biological matrices. J Chromatogr A. 2015; 1426:33–47. doi:10.1016/j.chroma.2015.11.040 ↩
66TakeuchiT, MinatoY, TakaseM, ShinmoriH. Molecularly imprinted polymers with halogen bonding-based molecular recognition sites. Tetrahedron Lett. 2005; 46(52):9025–9027. doi:10.1016/j.tetlet.2005.10.098 ↩
67BoulanouarS, MezzacheS, CombèsA, PichonV. Molecularly imprinted polymers for the determination of organophosphorus pesticides in complex samples. Talanta 2018; 176:465–478. doi:10.1016/j.talanta.2017.08.067 ↩
68AmbrosiniS, SerraM, ShindeS, SellergrenB, De LorenziE. Synthesis and chromatographic evaluation of molecularly imprinted polymers prepared by the substructure approach for the class-selective recognition of glucuronides. J Chromatogr A. 2011; 1218(39):6961–6969. doi:10.1016/j.chroma.2011.07.104 ↩
69YiL-X, FangR, ChenG-H. Molecularly imprinted solid-phase extraction in the analysis of agrochemicals. J Chromatogr Sci. 2013; 51(7):608–618. doi:10.1093/chromsci/bmt024 ↩
70ChenJ, TianY, ZhangY, XuF. Chemoselective probes serving as promising derivatization tools in targeted metabolomics research. J Anal Test. 2020; 4(3):175–182. doi:10.1007/s41664-020-00125-0 ↩
71CarlsonEE, CravattBF. Chemoselective probes for metabolite enrichment and profiling. Nat Methods. 2007; 4(5):429–435. doi:10.1038/nmeth1038 ↩
72GargN, ConwayLP, BalletC, et al Chemoselective probe containing a unique bioorthogonal cleavage site for investigation of gut microbiota metabolism. Angew Chem. 2018; 130(42):14001–14005. doi:10.1002/ange.201804828 ↩
73ConwayLP, GargN, LinW, VujasinovicM, LöhrJ-M, GlobischD. Chemoselective probe for detailed analysis of ketones and aldehydes produced by gut microbiota in human samples. Chem Commun (Camb). 2019; 55(62):9080–9083. doi:10.1039/C9CC04605D ↩
74ChungMK, RappaportSM, WheelockCE, et al Utilizing a Biology-Driven Approach to Map the Exposome in Health and Disease: An Essential Investment to Drive the Next Generation of Environmental Discovery. Environ Health Perspect. 2021 Aug; 129(8):85001. doi: 10.1289/EHP8327 ↩
75GysC, KovačičA, HuberC, LaiFY, HeathE, CovaciA. Suspect and untargeted screening of bisphenol S metabolites produced by in vitro human liver metabolism. Toxicol Lett. 2018; 295:115–123. doi:10.1016/j.toxlet.2018.05.034 ↩
76PlassmannMM, BrackW, KraussM. Extending analysis of environmental pollutants in human urine towards screening for suspected compounds. J Chromatogr A. 2015; 1394:18–25. doi:10.1016/j.chroma.2015.03.040 ↩
77CorreiaMSP, RaoM, BalletC, GlobischD. Coupled enzymatic treatment and mass spectrometric analysis for identification of glucuronidated metabolites in human samples. Chembiochem. 2019; 20(13):1678–1683. doi:10.1002/cbic.201900065 ↩
78BalletC, CorreiaMSP, ConwayLP, et al. New enzymatic and mass spectrometric methodology for the selective investigation of gut microbiota-derived metabolites. Chem Sci. 2018; 9(29):6233–6239. http://doi.org/10.1039/c8sc01502c 30090311 ↩
79BletsouAA, JeonJ, HollenderJ, ArchontakiE, ThomaidisNS. Targeted and non-targeted liquid chromatography-mass spectrometric workflows for identification of transformation products of emerging pollutants in the aquatic environment. Trends Anal Chem. 2015; 66:32–44. doi:10.1016/j.trac.2014.11.009 ↩
80JunkerJ, ChongI, KampF, et al Comparison of Strategies for the Determination of Sterol Sulfates via GC-MS Leading to a Novel Deconjugation-Derivatization Protocol. Molecules. 2019 Jun 26; 24(13):2353. doi: 10.3390/molecules24132353 ↩
81GongZ-G, HuJ, WuX, XuY-J. The recent developments in sample preparation for mass spectrometry-based metabolomics. Crit Rev Anal Chem. 2017; 47(4):325–331. doi:10.1080/10408347.2017.1289836 ↩
82WawrzyniakR, KosnowskaA, MacioszekS, BartoszewskiR, Jan MarkuszewskiM. New plasma preparation approach to enrich metabolome coverage in untargeted metabolomics: plasma protein bound hydrophobic metabolite release with proteinase K. Sci Rep. 2018 Jun 22; 8(1):9541. doi: 10.1038/s41598-018-27983-0 ↩
83LiuX, FangM, XuF, ChenD. Characterization of the binding of per- and poly-fluorinated substances to proteins: a methodological review. Trends Anal Chem. 2019; 116:177–185. doi:10.1016/j.trac.2019.05.017 ↩
84Environmental Protection Agency (EPA). SW-846 Test Method 3665A: Sulfuric acid/permanganate cleanup. 1996. https://www.epa.gov/hw-sw846/sw-846-test-method-3665a-sulfuric-acidpermanganate-cleanup. Accessed: 12.07.2021.https://www.epa.gov/hw-sw846/sw-846-test-method-3665a-sulfuric-acidpermanganate-cleanup ↩
85FiehnO. Metabolomics by gas chromatography–mass spectrometry: combined targeted and untargeted profiling. Curr Protoc Mol Biol. 2016; 114(1). doi:10.1002/0471142727.mb3004s114 ↩
86ZhaoS, LiL. Chemical derivatization in LC-MS-based metabolomics study. Trends Anal Chem. 2020; 131:115988. doi:10.1016/j.trac.2020.115988 ↩
87ZhaoS, LiH, HanW, ChanW, LiL. Metabolomic coverage of chemical-group-submetabolome analysis: group classification and four-channel chemical isotope labeling LC-MS. Anal Chem. 2019; 91(18):12108–12115. doi:10.1021/acs.analchem.9b03431 ↩
88HalketJM, WatermanD, PrzyborowskaAM, PatelRKP, FraserPD, BramleyPM. Chemical derivatization and mass spectral libraries in metabolic profiling by GC/MS and LC/MS/MS. J Exp Bot. 2005; 56(410):219–243. doi:10.1093/jxb/eri069 ↩
89KananiH, ChrysanthopoulosPK, KlapaMI. Standardizing GC–MS metabolomics. J Chromatogr B Analyt Technol Biomed Life Sci. 2008; 871(2):191–201. doi:10.1016/j.jchromb.2008.04.049 ↩
90BruheimP, KvitvangHFN, Villas-BoasSG. Stable isotope coded derivatizing reagents as internal standards in metabolite profiling. J Chromatogr A. 2013; 1296:196–203. doi:10.1016/j.chroma.2013.03.072 ↩
91BrownSD, RhodesDJ, PritchardBJ. A validated SPME-GC–MS method for simultaneous quantification of club drugs in human urine. Forensic Sci Int. 2007; 171(2-3):142–150. doi:10.1016/j.forsciint.2006.10.015 ↩
92JiaS, XuT, HuanT, et al Chemical isotope labeling exposome (CIL-EXPOSOME): one high-throughput platform for human urinary global exposome characterization. Environ Sci Technol. 2019; 53(9):5445–5453. doi:10.1021/acs.est.9b00285 ↩
93Pérez-FernándezV, Mainero RoccaL, TomaiP, FanaliS, GentiliA. Recent advancements and future trends in environmental analysis: Sample preparation, liquid chromatography and mass spectrometry. Anal Chim Acta. 2017 Aug 29; 983:9-41. doi: 10.1016/j.aca.2017.06.029 ↩
94MiggielsP, WoutersB, van WestenGJP, DubbelmanA-C, HankemeierT. Novel technologies for metabolomics: more for less. Trends Anal Chem. 2019; 120:115323. doi:10.1016/j.trac.2018.11.021 ↩
95HemmatiM, NixC, CrommenJ, ServaisA-C, FilletM. Benefits of microsampling and microextraction for metabolomics studies. Trends Anal Chem. 2020; 127:115899. doi:10.1016/j.trac.2020.115899 ↩
96OdoardiS, FisichellaM, RomoloFS, Strano-RossiS. High-throughput screening for new psychoactive substances (NPS) in whole blood by DLLME extraction and UHPLC–MS/MS analysis. J Chromatogr B Analyt Technol Biomed Life Sci. 2015; 1000:57–68. doi:10.1016/j.jchromb.2015.07.007 ↩
97Reyes-GarcésN, GionfriddoE, Gómez-RíosGA, et al Advances in solid phase microextraction and perspective on future directions. Anal Chem. 2018; 90(1):302–360. doi:10.1021/acs.analchem.7b04502 ↩
98VuckovicD, PawliszynJ. Systematic evaluation of solid-phase microextraction coatings for untargeted metabolomic profiling of biological fluids by liquid chromatography−mass spectrometry. Anal Chem. 2011; 83(6):1944–1954. doi:10.1021/ac102614v ↩
99MirnaghiFS, GoryńskiK, Rodriguez-LafuenteA, BoyacıE, BojkoB, PawliszynJ. Microextraction versus exhaustive extraction approaches for simultaneous analysis of compounds in wide range of polarity. J Chromatogr A. 2013; 1316:37–43. doi:10.1016/j.chroma.2013.09.084 ↩
100MousaviF, BojkoB, PawliszynJ. Development of high throughput 96-blade solid phase microextraction-liquid chromatrography-mass spectrometry protocol for metabolomics. Analytica Chimica Acta. 2015; 892:95–104. doi:10.1016/j.aca.2015.08.016 ↩
101JarochK, GoryńskaPZ, GoryńskiK, StefańskiT, BojkoB. Untargeted screening of phase I metabolism of combretastatin A4 by multi-tool analysis. Talanta 2018; 182:22–31. doi:10.1016/j.talanta.2018.01.051
102AllanIJ, HarmanC, RanneklevSB, ThomasKV, GrungM. Passive sampling for target and nontarget analyses of moderately polar and nonpolar substances in water: passive sampling for target and nontarget analyses. Environ Toxicol Chem. 2013; 32(8):1718–1726. doi:10.1002/etc.2260 ↩
103BessonneauV, IngsJ, McMasterM, et al In vivo microsampling to capture the elusive exposome. Sci Rep. 2017 Mar 7; 7:44038. doi: 10.1038/srep44038
104GrandyJJ, LashgariM, HeideHV, PooleJ, PawliszynJ. Introducing a mechanically robust SPME sampler for the on-site sampling and extraction of a wide range of untargeted pollutants in environmental waters. Environ Pollut. 2019; 252:825–834. doi:10.1016/j.envpol.2019.06.013 ↩
105ChallisJK, AlmirallXO, HelmPA, WongCS. Performance of the organic-diffusive gradients in thin-films passive sampler for measurement of target and suspect wastewater contaminants. Environ Pollut. 2020; 261:114092. doi: 10.1016/j.envpol.2020.114092
106VincentB, JenniferI, MarkM, et al In vivo tissue sampling using solid-phase microextraction for non-lethal exposome-wide association study of CYP1A1 induction in Catostomus commersonii. Environ Res. 2016; 151:216–223. doi:10.1016/j.envres.2016.07.006
107RoszkowskaA, YuM, BessonneauV, et al In vivo solid-phase microextraction sampling combined with metabolomics and toxicological studies for the non-lethal monitoring of the exposome in fish tissue. Environ Pollut. 2019; 249:109–115. doi:10.1016/j.envpol.2019.03.024
108BaoL-J, ZengEY. Passive sampling techniques for sensing freely dissolved hydrophobic organic chemicals in sediment porewater. Trends Anal Chem. 2011; 30(9):1422–1428. doi:10.1016/j.trac.2011.05.004 ↩
109OuyangG, PawliszynJ. Recent developments in SPME for on-site analysis and monitoring. Trends Anal Chem. 2006; 25(7):692–703. doi:10.1016/j.trac.2006.05.005 ↩
110ReichenbergF, MayerP. Two complementary sides of bioavailability: accessibility and chemical activity of organic contaminants in sediments and soils. Environ Toxicol Chem. 2006; 25(5):1239–1245. DOI: 10.1897/05-458r.1 ↩
111MayerP, TollsJ, HermensJL, MackayD. Equilibrium sampling devices. Environ Sci Technol. 2003 May 1; 37(9):184A-191A. doi: 10.1021/es032433i ↩
112WangS, OakesKD, BraggLM, PawliszynJ, DixonG, ServosMR. Validation and use of in vivo solid phase micro-extraction (SPME) for the detection of emerging contaminants in fish. Chemosphere 2011; 85(9):1472–1480. doi:10.1016/j.chemosphere.2011.08.035 ↩
113DohertyBT, KoelmelJP, LinEZ, RomanoME, Godri PollittKJ. Use of exposomic methods incorporating sensors in environmental epidemiology [published online ahead of print]. Curr Environ Health Rep. 2021; 8(1):34–41. doi:10.1007/s40572-021-00306-8 ↩
114GionfriddoE, BoyacıE, PawliszynJ. New generation of solid-phase microextraction coatings for complementary separation approaches: a step toward comprehensive metabolomics and multiresidue analyses in complex matrices. Anal Chem. 2017; 89(7):4046–4054. doi:10.1021/acs.analchem.6b04690 ↩
115VasiljevicT, SinghV, PawliszynJ. Miniaturized SPME tips directly coupled to mass spectrometry for targeted determination and untargeted profiling of small samples. Talanta 2019; 199:689–697. doi:10.1016/j.talanta.2019.03.025 ↩
116MousaviF. Optimization of SPME coating characteristics for metabolomics and targeted analysis with LC/MS. Doctoral thesis; University of Waterloo, Canada. . 2015. http://hdl.handle.net/10012/9696. Accessed: 08.02.2021.http://hdl.handle.net/10012/9696 ↩
117XuJ, HuangS, WuR, et al Bioinspired polydopamine sheathed nanofibers for high-efficient in vivo solid-phase microextraction of pharmaceuticals in fish muscle. Anal Chem. 2015; 87(6):3453–3459. doi:10.1021/ac5048357 ↩
118RiboniF, BianchiC. Recent advances in in vivo SPME sampling. Separations 2020; 7(1):6. doi:10.3390/separations7010006 ↩
119FedrizziB, CarlinS, FranceschiP, et al D-optimal design of an untargeted HS-SPME-GC-TOF metabolite profiling method. Analyst. 2012; 137(16):3725–3731. doi:10.1039/c2an16309h ↩
120AraújoAM, MoreiraN, LimaAR, et al Analysis of extracellular metabolome by HS-SPME/GC–MS: optimization and application in a pilot study to evaluate galactosamine-induced hepatotoxicity. Toxicol Lett. 2018; 295:22–31. doi:10.1016/j.toxlet.2018.05.028
121PurcaroG, StefanutoP-H, FranchinaFA, et al SPME-GC×GC-TOF MS fingerprint of virally-infected cell culture: sample preparation optimization and data processing evaluation. Anal Chim Acta. 2018; 1027:158–167. doi:10.1016/j.aca.2018.03.037
122VitaleCM, Knudsmark SjøholmK, Di GuardoA, MayerP. Accelerated equilibrium sampling of hydrophobic organic chemicals in solid matrices: A proof of concept on how to reach equilibrium for PCBs within 1 day. Chemosphere 2019; 237:124537.doi:10.1016/j.chemosphere.2019.124537
123PriceEJ, PalátJ, CoufalikováK, et al Open, high-resolution EI+ spectral library of anthropogenic compounds. Front Public Health. 2021; 9:622558. doi:10.3389/fpubh.2021.622558 ↩
124StettinD, PoulinRX, PohnertG. Metabolomics Benefits from Orbitrap GC-MS-Comparison of Low- and High-Resolution GC-MS. Metabolites. 2020 Apr 4; 10(4):143. doi: 10.3390/metabo10040143 ↩
125RavindraK, DirtuAC, CovaciA. Low-pressure gas chromatography: recent trends and developments. Trends Anal Chem. 2008; 27(4):291–303. doi:10.1016/j.trac.2008.02.003 ↩
126Lehotay, S.J., de Zeeuw, J., Sapozhnikova, Y., Michlig, N., Hepner, J.R., Konschnik, J.D., 2020. There is no time to waste: Low-pressure gas chromatography–mass spectrometry is a proven solution for fast, sensitive, and robust GC–MS analysis. LC-GC North Am. 2020;38(8), 457–466. https://cdn.sanity.io/files/0vv8moc6/chroma/f562afa926baa1c61c935270836a3374f513bb14.pdf/LCGC_NAmerica_August2020%20(1).pdf. Accessed: 12.07.2021.https://cdn.sanity.io/files/0vv8moc6/chroma/f562afa926baa1c61c935270836a3374f513bb14.pdf/LCGC_NAmerica_August2020%20(1).pdf ↩
127ŠpánikI, MachyňákováA. Recent applications of gas chromatography with high-resolution mass spectrometry. J Sep Sci. 2018; 41(1):163–179. doi:10.1002/jssc.201701016 ↩
128SeeleyJV, SeeleySK. Multidimensional gas chromatography: fundamental advances and new applications. Anal Chem. 2013; 85(2):557–578. doi:10.1021/ac303195u ↩
129Higgins KepplerEA, JenkinsCL, DavisTJ, BeanHD. Advances in the application of comprehensive two-dimensional gas chromatography in metabolomics. Trends Analyt Chem. 2018; 109:275–286. doi:10.1016/j.trac.2018.10.015 ↩
130AspromonteJ, WolfsK, AdamsE. Current application and potential use of GC × GC in the pharmaceutical and biomedical field. J Pharm Biomed Anal. 2019 Nov 30; 176:112817. doi: 10.1016/j.jpba.2019.112817 ↩
131MuscaluAM, GóreckiT. Comprehensive two-dimensional gas chromatography in environmental analysis. Trends Anal Chem. 2018; 106:225–245. doi:10.1016/j.trac.2018.07.001 ↩
132BahaghighatHD, FreyeCE, SynovecRE. Recent advances in modulator technology for comprehensive two dimensional gas chromatography. Trends Anal Chem. 2019; 113:379–391. doi:10.1016/j.trac.2018.04.016 ↩
133Margolin ErenKJ, ElkabetsO, AmiravA. A comparison of electron ionization mass spectra obtained at 70 eV, low electron energies, and with cold EI and their NIST library identification probabilities. J Mass Spectrom. 2020 Dec; 55(12):e4646. doi: 10.1002/jms.4646 ↩
134HongS, KangW, SuY, GuoY. Analysis of trace-level volatile compounds in fresh turf crop (Lolium perenne L.) by gas chromatography quadrupole time-of-flight mass spectrometry. Chin J Chem. 2013; 31(10):1329–1335. doi:10.1002/cjoc.201300414 ↩
135OmerE, BichonE, HutinetS, et al Toward the characterisation of non-intentionally added substances migrating from polyester-polyurethane lacquers by comprehensive gas chromatography-mass spectrometry technologies. J Chromatogr A. 2019; 1601:327–334. doi:10.1016/j.chroma.2019.05.024 ↩
136AmiravA, GordinA, PoliakM, FialkovAB. Gas chromatography-mass spectrometry with supersonic molecular beams. J Mass Spectrom. 2008; 43(2):141–163. doi:10.1002/jms.1380 ↩
137AmiravA, KeshetU, DanonA. Soft cold EI - approaching molecular ion only with electron ionization: soft cold EI. Rapid Commun Mass Spectrom. 2015; 29(21):1954–1960. doi:10.1002/rcm.7305 ↩
138TsizinS, BokkaR, KeshetU, et al Comparison of electrospray LC–MS, LC–MS with cold EI and GC–MS with cold EI for sample identification. Int J Mass Spectrom. 2017; 422:119–125. doi:10.1016/j.ijms.2017.09.006 ↩
139MisraBB, OlivierM. High resolution GC-orbitrap-MS metabolomics using both electron ionization and chemical ionization for analysis of human plasma. J Proteome Res. 2020; 19(7):2717–2731. doi:10.1021/acs.jproteome.9b00774 ↩
140HaulerC, VetterW. A non-targeted gas chromatography/electron capture negative ionization mass spectrometry selected ion monitoring screening method for polyhalogenated compounds in environmental samples: non-targeted GC/ECNI-MS-SIM screening of polyhalogenated compounds. Rapid Commun Mass Spectrom. 2015; 29(7):619–628. doi:10.1002/rcm.7143 ↩
141MazurDM, DetenchukEA, SosnovaAA, ArtaevVB, LebedevAT. GC-HRMS with complementary ionization techniques for target and non-target screening for chemical exposure: expanding the insights of the air pollution markers in Moscow snow. Sci Total Environ. 2021; 761:144506. doi:10.1016/j.scitotenv.2020.144506
142XiaoX-Y, McCalleyDV, McEvoyJ. Analysis of estrogens in river water and effluents using solid-phase extraction and gas chromatography–negative chemical ionisation mass spectrometry of the pentafluorobenzoyl derivatives. J Chromatogr A. 2001; 923(1-2):195–204. doi:10.1016/S0021-9673(01)00955-4
143RochatB. From targeted quantification to untargeted metabolomics: why LC-high-resolution-MS will become a key instrument in clinical labs. Trends Anal Chem. 2016; 84:151–164. doi:10.1016/j.trac.2016.02.009 ↩
144López-RuizR, Romero-GonzálezR, Garrido FrenichA. Ultrahigh-pressure liquid chromatography-mass spectrometry: an overview of the last decade. TrAC Trends in Analytical Chemistry. 2019; 118:170–181. doi:10.1016/j.trac.2019.05.044 ↩
145HayesR, AhmedA, EdgeT, ZhangH. Core–shell particles: preparation, fundamentals and applications in high performance liquid chromatography. J Chromatogr A. 2014; 1357:36–52. doi:10.1016/j.chroma.2014.05.010 ↩
146PeriatA, GuillarmeD, VeutheyJ-L, et al Optimized selection of liquid chromatography conditions for wide range analysis of natural compounds. J Chromatogr A. 2017; 1504:91–104. doi:10.1016/j.chroma.2017.05.024 ↩
147ContrepoisK, JiangL, SnyderM. optimized analytical procedures for the untargeted metabolomic profiling of human urine and plasma by combining hydrophilic interaction (HILIC) and reverse-phase liquid chromatography (RPLC)–mass spectrometry. Mol Cell Proteomics. 2015; 14(6):1684–1695. doi:10.1074/mcp.M114.046508 ↩
148ZhangT, CreekDJ, BarrettMP, BlackburnG, WatsonDG. Evaluation of coupling reversed phase, aqueous normal phase, and hydrophilic interaction liquid chromatography with orbitrap mass spectrometry for metabolomic studies of human urine. Anal Chem. 2012; 84(4):1994–2001. doi:10.1021/ac2030738 ↩
149NaserFJ, MahieuNG, WangL, SpaldingJL, JohnsonSL, PattiGJ. Two complementary reversed-phase separations for comprehensive coverage of the semipolar and nonpolar metabolome. Anal Bioanal Chem. 2018; 410(4):1287–1297. doi:10.1007/s00216-017-0768-x ↩
150Narváez-RivasM, ZhangQ. Comprehensive untargeted lipidomic analysis using core–shell C30 particle column and high field orbitrap mass spectrometer. J Chromatogr A. 2016; 1440:123–134. doi:10.1016/j.chroma.2016.02.054 ↩
151RimmerCA, SanderLC, WiseSA. Selectivity of long chain stationary phases in reversed phase liquid chromatography. Anal Bioanal Chem. 2005; 382(3):698–707. doi:10.1007/s00216-004-2858-9 ↩
152BapiroTE, RichardsFM, JodrellDI. Understanding the complexity of porous graphitic carbon (PGC) chromatography: modulation of mobile-stationary phase interactions overcomes loss of retention and reduces variability. Anal Chem. 2016; 88(12):6190–6194. doi:10.1021/acs.analchem.6b01167 ↩
153GoncharovaEN, StatkusMA, TsizinGI, ZolotovYA. Porous graphitized carbon for the separation and preconcentration of hydrophilic substances. J Anal Chem. 2020; 75(4):423–442. doi:10.1134/S1061934820040036 ↩
154RamplerE, AbieadYE, SchoenyH, et al Recurrent topics in mass spectrometry-based metabolomics and lipidomics—standardization, coverage, and throughput. Anal Chem. 2021; 93(1):519–545. doi:10.1021/acs.analchem.0c04698 ↩
155YangQ, ShiX, WangY, et al Urinary metabonomic study of lung cancer by a fully automatic hyphenated hydrophilic interaction/RPLC‐MS system. J Sep Sci. 2010; 33(10):1495–1503. doi:10.1002/jssc.200900798 ↩
156ChalcraftKR, McCarryBE. Tandem LC columns for the simultaneous retention of polar and nonpolar molecules in comprehensive metabolomics analysis: Liquid Chromatography. J Sep Sci. 2013; 36(21-22):3478–3485. doi:10.1002/jssc.201300779 ↩
157HaggartyJ, OppermannM, DalbyMJ, et al Serially coupling hydrophobic interaction and reversed-phase chromatography with simultaneous gradients provides greater coverage of the metabolome. Metabolomics. 2015; 11(5):1465–1470. doi:10.1007/s11306-014-0770-7
158Mejía-CarmonaK, Soares da Silva BuratoJ, BorsattoJVB, de ToffoliAL, LancFM. Miniaturization of liquid chromatography coupled to mass spectrometry. Trends Anal Chem. 2020; 122:115735. doi:10.1016/j.trac.2019.115735 ↩
159Vargas MedinaDA, MacielEVS, de ToffoliAL, LancFM. Miniaturization of liquid chromatography coupled to mass spectrometry. Trends Anal Chem. 2020; 128:115910. doi: 10.1016/j.trac.2020.115910
160Vargas MedinaDA, MacielEVS, LancFM. Miniaturization of liquid chromatography coupled to mass spectrometry. 3. Achievements on chip-based LC–MS devices. Trends Anal Chem. 2020; 131:116003. doi:10.1016/j.trac.2020.116003
161LenzEM, WilliamsRE, SidawayJ, et al The application of microbore UPLC/oa-TOF-MS and 1H NMR spectroscopy to the metabonomic analysis of rat urine following the intravenous administration of pravastatin. J Pharm Biomed Anal. 2007; 44(4):845–852. doi:10.1016/j.jpba.2007.04.035 ↩
162SaitoY, JinnoK, GreibrokkT. Capillary columns in liquid chromatography: between conventional columns and microchips. J Sep Sci. 2004; 27(17-18):1379–1390. doi:10.1002/jssc.200401902 ↩
163MetzTO, PageJS, BakerES, et al High-resolution separations and improved ion production and transmission in metabolomics. Trends Analyt Chem. 2008; 27(3):205–214. doi:10.1016/j.trac.2007.11.003 ↩
164GrayN, LewisMR, PlumbRS, WilsonID, NicholsonJK. High-throughput microbore UPLC–MS metabolic phenotyping of urine for large-scale epidemiology studies. J Proteome Res. 2015; 14(6):2714–2721. doi:10.1021/acs.jproteome.5b00203 ↩
165RainvillePD, SimeoneJL, McCarthySM, SmithNW, CowanD, PlumbRS. Investigation of microbore UPLC and nontraditional mobile phase compositions for bioanalytical LC–MS/MS. Bioanalysis 2012; 4(11):1287–1297. doi:10.4155/bio.12.78 ↩
166ChetwyndAJ, DavidA. A review of nanoscale LC-ESI for metabolomics and its potential to enhance the metabolome coverage. Talanta 2018; 182:380–390. doi:10.1016/j.talanta.2018.01.084 ↩
167GrangerJ, PlumbR, Castro-PerezJ, WilsonID. Metabonomic studies comparing capillary and conventional HPLC-oa-TOF MS for the analysis of urine from zucker obese rats. Chroma. 2005; 61(7-8):375–380. doi:10.1365/s10337-005-0523-x ↩
168ZhangJ, ShouW, OguraT, LiS, WellerH. Optimization of microflow LC–MS/MS and its utility in quantitative discovery bioanalysis. Bioanalysis 2019; 11(11):1117–1127. doi:10.4155/bio-2019-0076 ↩
169WilmM, MannM. Analytical properties of the nanoelectrospray ion source. Anal Chem. 1996; 68(1):1–8. doi:10.1021/ac9509519 ↩
170ChetwyndAJ, DavidA, HillEM, Abdul-SadaA. Evaluation of analytical performance and reliability of direct nanoLC-nanoESI-high resolution mass spectrometry for profiling the (xeno)metabolome: nanoUHPLC-nanoESI-TOFMS for (xeno)metabolomics. J Mass Spectrom. 2014; 49(10):1063–1069. doi:10.1002/jms.3426 ↩
171WilsonSR, VehusT, BergHS, LundanesE. Nano-LC in proteomics: recent advances and approaches. Bioanalysis 2015; 7(14):1799–1815. doi:10.4155/bio.15.92 ↩
172NysG, CobraivilleG, FilletM. Multidimensional performance assessment of micro pillar array column chromatography combined to ion mobility-mass spectrometry for proteome research. Anal Chim Acta. 2019; 1086:1–13. doi:10.1016/j.aca.2019.08.068 ↩
173Choi, K., Boyaci, E., Kim, J., Seale, B., Barrera-Arbelaez, L., Pawliszyn, J., Wheeler, A.R. A digital microfluidic interface between solid-phase microextraction and liquid chromatography-mass spectrometry. J. Chromatogr. A. 2016;1444, 1–7. doi:10.1016/j.chroma.2016.03.029 ↩
174PetrieB, YoudanJ, BardenR, Kasprzyk-HordernB. Multi-residue analysis of 90 emerging contaminants in liquid and solid environmental matrices by ultra-high-performance liquid chromatography tandem mass spectrometry. J Chromatogr A. 2016; 1431:64–78. doi:10.1016/j.chroma.2015.12.036 ↩
175PreindlK, BraunD, AichingerG, et al A generic liquid chromatography−tandem mass spectrometry exposome method for the determination of xenoestrogens in biological matrices. Anal Chem. 2019; 91(17):11334–11342. doi:10.1021/acs.analchem.9b02446 ↩
176NarduzziL, RoyerA-L, BichonE, et al Ammonium fluoride as suitable additive for HILIC-based LC-HRMS metabolomics. Metabolites 2019; 9(12):292. doi:10.3390/metabo9120292 ↩
177ZhangX, ClausenMR, ZhaoX, ZhengH, BertramHC. Enhancing the power of liquid chromatography–mass spectrometry-based urine metabolomics in negative ion mode by optimization of the additive. Anal Chem. 2012; 84(18):7785–7792. doi:10.1021/ac3013835 ↩
178CreydtM, FischerM. Plant metabolomics: maximizing metabolome coverage by optimizing mobile phase additives for nontargeted mass spectrometry in positive and negative electrospray ionization mode. Anal Chem. 2017; 89(19):10474–10486. doi:10.1021/acs.analchem.7b02592 ↩
179LuW, XingX, WangL, et al Improved annotation of untargeted metabolomics data through buffer modifications that shift adduct mass and intensity. Anal Chem. 2020; 92(17):11573–11581. doi:10.1021/acs.analchem.0c00985 ↩
180YanB, ZhaoJ, BrownJS, BlackwellJ, CarrPW. High-temperature ultrafast liquid chromatography. Anal Chem. 2000; 72(6):1253–1262. doi:10.1021/ac991008y ↩
181GuillarmeD, RutaJ, RudazS, VeutheyJ-L. New trends in fast and high-resolution liquid chromatography: a critical comparison of existing approaches. Anal Bioanal Chem. 2010; 397(3):1069–1082. doi:10.1007/s00216-009-3305-8 ↩
182RainvillePD, TheodoridisG, PlumbRS, WilsonID. Advances in liquid chromatography coupled to mass spectrometry for metabolic phenotyping. Trends Anal Chem. 2014; 61:181–191. doi:10.1016/j.trac.2014.06.005 ↩
183PereiraL, AspeyS, WoodrufM, RitchieH. Advantages of using ultra high temperature in high throughput LC and LC/MS [published online ahead of print]; 2021. http://www.thermo.com.cn/Resources/200802/productPDF_25323.pdf. Accessed: 12.07.2021.http://www.thermo.com.cn/Resources/200802/productPDF_25323.pdf ↩
184PlumbRS, RainvilleP, SmithBW, et al Generation of ultrahigh peak capacity LC separations via elevated temperatures and high linear mobile-phase velocities. Anal Chem. 2006; 78(20):7278–7283. doi:10.1021/ac060935j ↩
185TheodoridisG, GikaHG, WilsonID. LC-MS-based methodology for global metabolite profiling in metabonomics/metabolomics. Trends Anal Chem. 2008; 27(3):251–260. doi:10.1016/j.trac.2008.01.008 ↩
186NguyenDT-T, GuillarmeD, HeinischS, et al High throughput liquid chromatography with sub-2μm particles at high pressure and high temperature. J Chromatogr A. 2007; 1167(1):76–84. doi:10.1016/j.chroma.2007.08.032 ↩
187PlumbR, MazzeoJR, GrumbachES, et al The application of small porous particles, high temperatures, and high pressures to generate very high resolution LC and LC/MS separations. J Sep Sci. 2007; 30(8):1158–1166. doi:10.1002/jssc.200600492 ↩
188GikaHG, TheodoridisG, ExtanceJ, EdgeAM, WilsonID. High temperature-ultra performance liquid chromatography–mass spectrometry for the metabonomic analysis of Zucker rat urine. J Chromatogr B Analyt Technol Biomed Life Sci. 2008; 871(2):279–287. doi:10.1016/j.jchromb.2008.04.020 ↩
189NordströmA, WantE, NorthenT, LehtiöJ, SiuzdakG. Multiple ionization mass spectrometry strategy used to reveal the complexity of metabolomics. Anal Chem. 2008; 80(2):421–429. doi:10.1021/ac701982e ↩
190LeiZ, HuhmanDV, SumnerLW. Mass spectrometry strategies in metabolomics. J Biol Chem. 2011; 286(29):25435–25442. doi:10.1074/jbc.R111.238691 ↩
191WilliamsDK, McAlisterGC, GoodDM, CoonJJ, MuddimanDC. Dual electrospray ion source for electron-transfer dissociation on a hybrid linear ion trap−orbitrap mass spectrometer. Anal Chem. 2007; 79(20):7916–7919. doi:10.1021/ac071444h ↩
192CajkaT, FiehnO. Toward merging untargeted and targeted methods in mass spectrometry-based metabolomics and lipidomics. Anal Chem. 2016; 88(1):524–545. doi:10.1021/acs.analchem.5b04491 ↩
193SchiewekR, LorenzM, GieseR, et al Development of a multipurpose ion source for LC-MS and GC-API MS. Anal Bioanal Chem. 2008; 392(1-2):87–96. doi:10.1007/s00216-008-2255-x ↩
194BrechtD, UteschilF, SchmitzOJ. Development of a fast-switching dual (ESI/APCI) ionization source for liquid chromatography/mass spectrometry. Rapid Commun Mass Spectrom. 2020; 34(17):e8845. doi:10.1002/rcm.8845 ↩
195TrimpinS. Novel ionization processes for use in mass spectrometry: ‘squeezing’ nonvolatile analyte ions from crystals and droplets. Rapid Commun Mass Spectrom. 2019; 33 Suppl 3(S3):96–120. doi:10.1002/rcm.8269 ↩
196FennerMA, ChakrabartyS, WangB, et al An LC/MS method providing improved sensitivity: electrospray ionization inlet. Anal Chem. 2017; 89(9):4798–4802. doi:10.1021/acs.analchem.6b05172 ↩
197TermopoliV, FamigliniG, PalmaP, PiergiovanniM, CappielloA. Atmospheric pressure vaporization mechanism for coupling a liquid phase with electron ionization mass spectrometry. Anal Chem. 2017; 89(3):2049–2056. doi:10.1021/acs.analchem.6b04646 ↩
198TermopoliV, FamigliniG, PalmaP, et al Evaluation of a liquid electron ionization liquid chromatography–mass spectrometry interface. J Chromatogr A. 2019; 1591:120–130. doi:10.1016/j.chroma.2019.01.034
199TsizinS, FialkovAB, AmiravA. Electron ionization mass spectrometry for both liquid and gas chromatography in one system without the need for hardware adjustments. J Am Soc Mass Spectrom. 2020; 31(8):1713–1721. doi:10.1021/jasms.0c00136
200DualdeP, CoscollàC, PastorA, YusàV. Optimization of resolving power, fragmentation, and mass calibration in an orbitrap spectrometer for analysis of 24 pesticide metabolites in urine. Int J Anal Chem. 2019; 2019:1917369–1917312. doi:10.1155/2019/1917369 ↩
201Xue, J., Domingo-Almenara, X., Guijas, C., Palermo, A., Rinschen, M.M., Isbell, J., Benton, H.P., Siuzdak, G. Enhanced in-Source Fragmentation Annotation Enables Novel Data Independent Acquisition and Autonomous METLIN Molecular Identification. Anal. Chem. 2020; 92, 6051–6059. doi:10.1021/acs.analchem.0c00409 ↩
202HutchinsPD, RussellJD, CoonJJ. Accelerating lipidomic method development through in silico simulation. Anal Chem. 2019; 91(15):9698–9706. doi:10.1021/acs.analchem.9b01234 ↩
203WandyJ, DaviesV, van der HooftJ, WeidtS, DalyR, RogersS. In silico optimization of mass spectrometry fragmentation strategies in metabolomics. Metabolites 2019; 9(10):219. doi: 10.3390/metabo9100219 ↩
204DecaesteckerTN, Vande CasteeleSR, WallemacqPE, Van PeteghemCH, DeforeDL, Van BocxlaerJF. Information-dependent acquisition-mediated LC−MS/MS screening procedure with semiquantitative potential. Anal Chem. 2004; 76(21):6365–6373. doi:10.1021/ac0492315 ↩
205ChoK, Schwaiger-HaberM, NaserFJ, StancliffeE, SindelarM, PattiGJ. Targeting unique biological signals on the fly to improve MS/MS coverage and identification efficiency in metabolomics. Anal ChimActa. 2021; 1149:338210. doi:10.1016/j.aca.2021.338210
206BroecklingCD, HoyesE, RichardsonK, BrownJM, PrenniJE. Comprehensive tandem-mass-spectrometry coverage of complex samples enabled by data-set-dependent acquisition. Anal Chem. 2018; 90(13):8020–8027. doi:10.1021/acs.analchem.8b00929
207KongSW, Hernandez-FerrerC. Assessment of coverage for endogenous metabolites and exogenous chemical compounds using an untargeted metabolomics platform. Biocomputing 2020:587–598. doi:10.1142/9789811215636_0052 ↩
208The Standard Metabolic Reporting Structures working group. Summary recommendations for standardization and reporting of metabolic analyses. Nat Biotechnol. 2005; 23(7):833–838. doi:10.1038/nbt0705-833 ↩
209SumnerLW, AmbergA, BarrettD, et al Proposed minimum reporting standards for chemical analysis: Chemical Analysis Working Group (CAWG) metabolomics standards initiative (MSI). Metabolomics. 2007; 3(3):211–221. doi:10.1007/s11306-007-0082-2 ↩
210SchulzeB, JeonY, KaserzonS, et al An assessment of quality assurance/quality control efforts in high resolution mass spectrometry non-target workflows for analysis of environmental samples. Trends Anal Chem. 2020; 133:116063. doi:10.1016/j.trac.2020.116063 ↩
211Caballero-CaseroN, BelovaL, VervlietP, et al Towards harmonised criteria in quality assurance and quality control of suspect and non-target LC-HRMS analytical workflows for screening of emerging contaminants in human biomonitoring. Trends Anal Chem. 2021; 136:116201. doi:10.1016/j.trac.2021.116201 ↩
212Rocca-SerraP, SalekRM, AritaM, et al Data standards can boost metabolomics research, and if there is a will, there is a way. Metabolomics. 2016; 12(1):14. doi:10.1007/s11306-015-0879-3 ↩
213SpicerRA, SalekR, SteinbeckC. A decade after the metabolomics standards initiative it’s time for a revision. Sci Data. 2017; 4(1):170138. doi:10.1038/sdata.2017.138 ↩
214SpicerRA, SalekR, SteinbeckC. Compliance with minimum information guidelines in public metabolomics repositories. Sci Data. 2017; 4(1):170137. doi:10.1038/sdata.2017.137 ↩